将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

编辑 | 萝卜皮
非编码 RNA 在各种生物功能中发挥着调控作用,并且与人类健康、药物设计等领域息息相关。
了解功能的机械机制需要三级结构信息,然而,通过实验确定 RNA 三维结构成本高昂且耗时,导致 RNA 序列和结构数据之间存在巨大差距。
为了应对这一挑战,普渡大学(Purdue University West Lafayette)的研究人员开发了 NuFold 来准确预测 RNA 三级结构。
NuFold 是一个深度神经网络,针对输入序列的输出结构进行端到端训练;它采用了碱基中心表示法,可实现核糖环的灵活构象。
NuFold 在构建正确的 RNA 局部几何结构方面表现出特殊优势。此外,NuFold 还能够通过链接输入序列来预测 RNA 的多聚体复合结构。
该研究以「NuFold: end-to-end approach for RNA tertiary structure prediction with flexible nucleobase center representation」为题,于 2025 年 1 月 21 日发布在《Nature Communications》。

核糖核酸 (RNA) 是生物体的基本分子。除了作为转录中使用的信使 RNA 发挥核心作用外,RNA 分子还以非编码 RNA (ncRNA) 的形式发挥各种生物学功能,它们参与基因调控和修饰等功能。
RNACentral 数据库目前包含超过三千万个 ncRNA 序列。ncRNA 在药物设计中也备受关注,因为新药可能被设计成抑制或模仿功能性 RNA 的活性。
在最新的研究中,普渡大学的研究团队开发了一种使用端到端深度网络架构的从头 RNA 结构预测方法 NuFold。该方法采用目标 RNA 序列,并通过经过全面训练的单个网络生成三级结构模型。
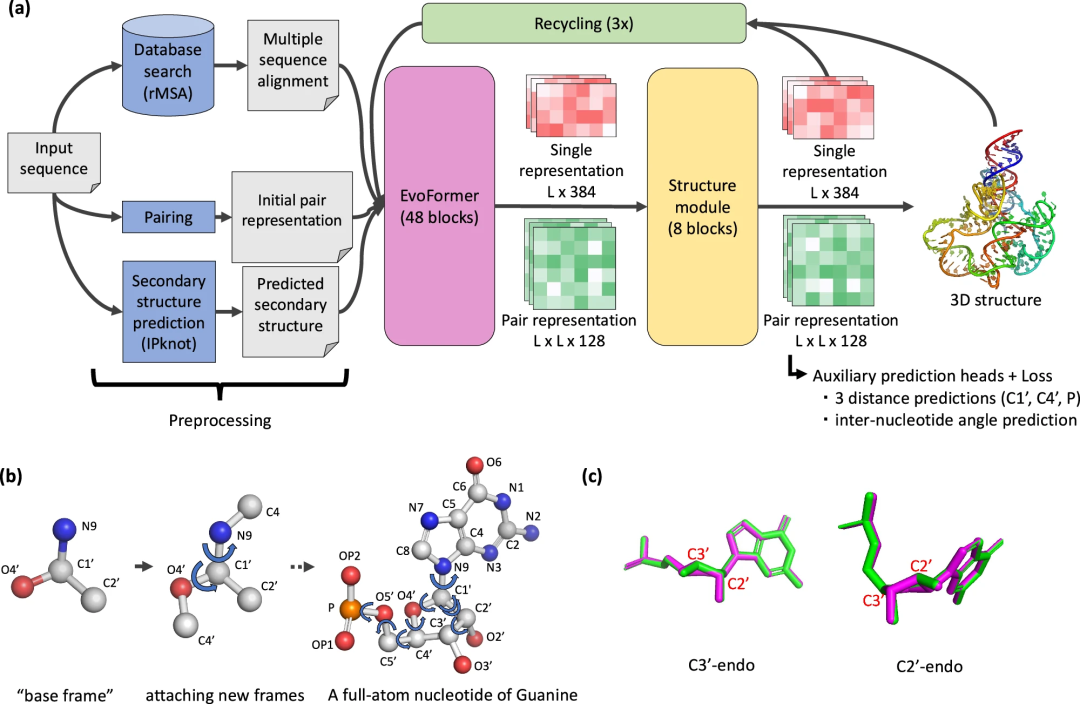
图示:NuFold 概述。(来源:论文)
该网络架构基于 AlphaFold2(AF2),这是一种蛋白质结构预测方法,在 2020 年的 CASP14(结构预测关键评估)中取得了出色的表现。
在 AF2 架构的基础上,研究人员进行了重大修改。这些修改包括调整核酸序列,以二级结构作为输入,修改预测 RNA 特定碱基间角度、距离和原子位置的方法,以及允许 RNA 结构表示具有完全的灵活性。
该团队的实现方法称为核碱基中心表示,可以优化核碱基所有可旋转键的角度。这种表示使他们能够重现碱基骨架中存在的任何灵活性,为精确的碱基构象建模奠定了基础。
与基于深度学习的方法相比,NuFold 是一种独特的端到端模型,它直接从 MSA 输出完整的原子模型并预测二级结构,这与许多其他深度学习模型不同,这些模型需要预测原子间距离和角度约束以用于后续的结构建模过程。
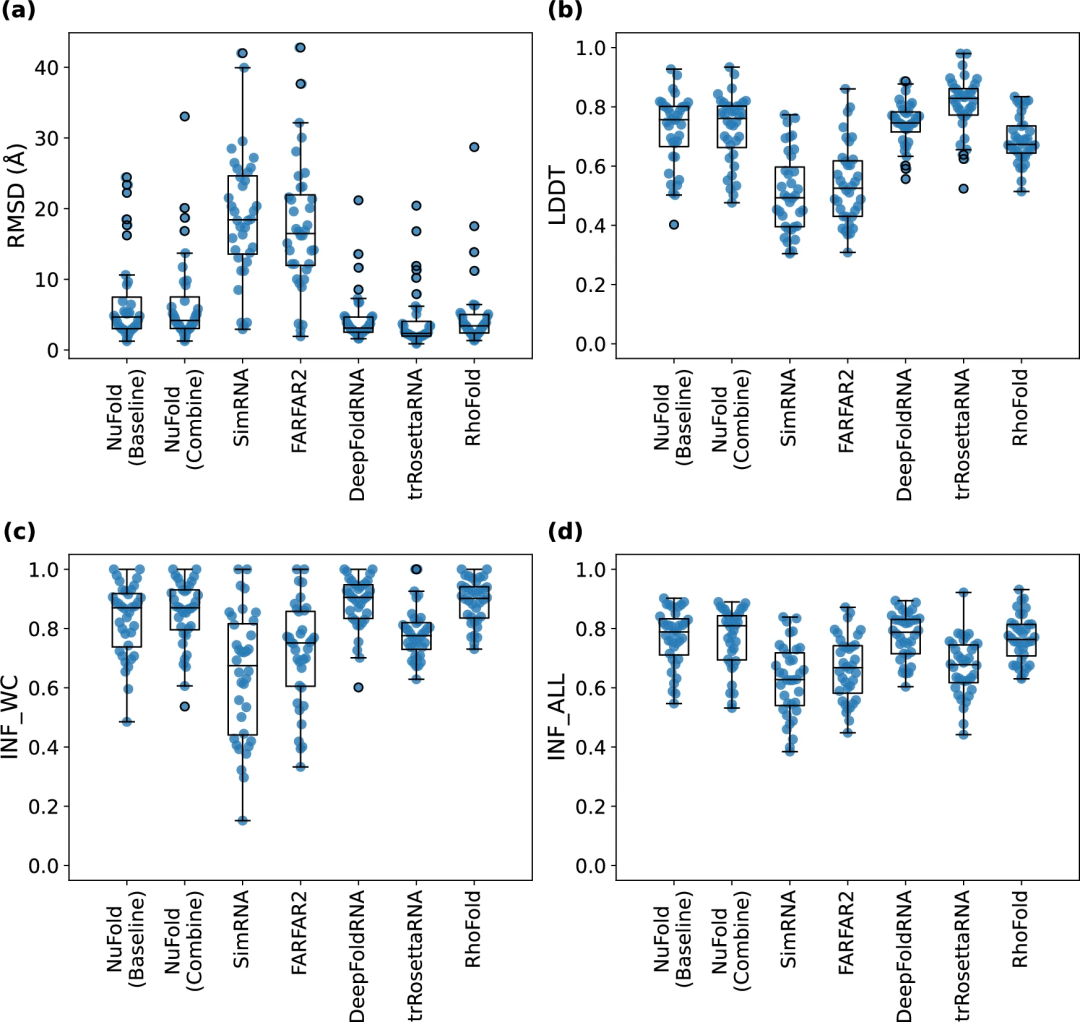
图示:基准结果用于比较预测方法。(来源:论文)
目前来讲,RhoFold 是唯一一个具有与 NuFold 类似架构的其他端到端模型。
这两个架构的明显差异在于,RhoFold 使用语言模型来处理输入的 MSA,而 NuFold 以类似于 AF2 的更直接的方式获取 MSA,并且 NuFold 将预测的二级结构信息作为另一个输入。
RNA 结构预测可能比蛋白质预测更困难,因为 RNA 分子更灵活,而且与蛋白质相比,可用的结构数据非常有限。该团队研究了几种克服这一困难的方法:为了增加训练数据的数量,研究人员采用了一种自提炼技术,将被认为足够准确的预测结构纳入训练集。
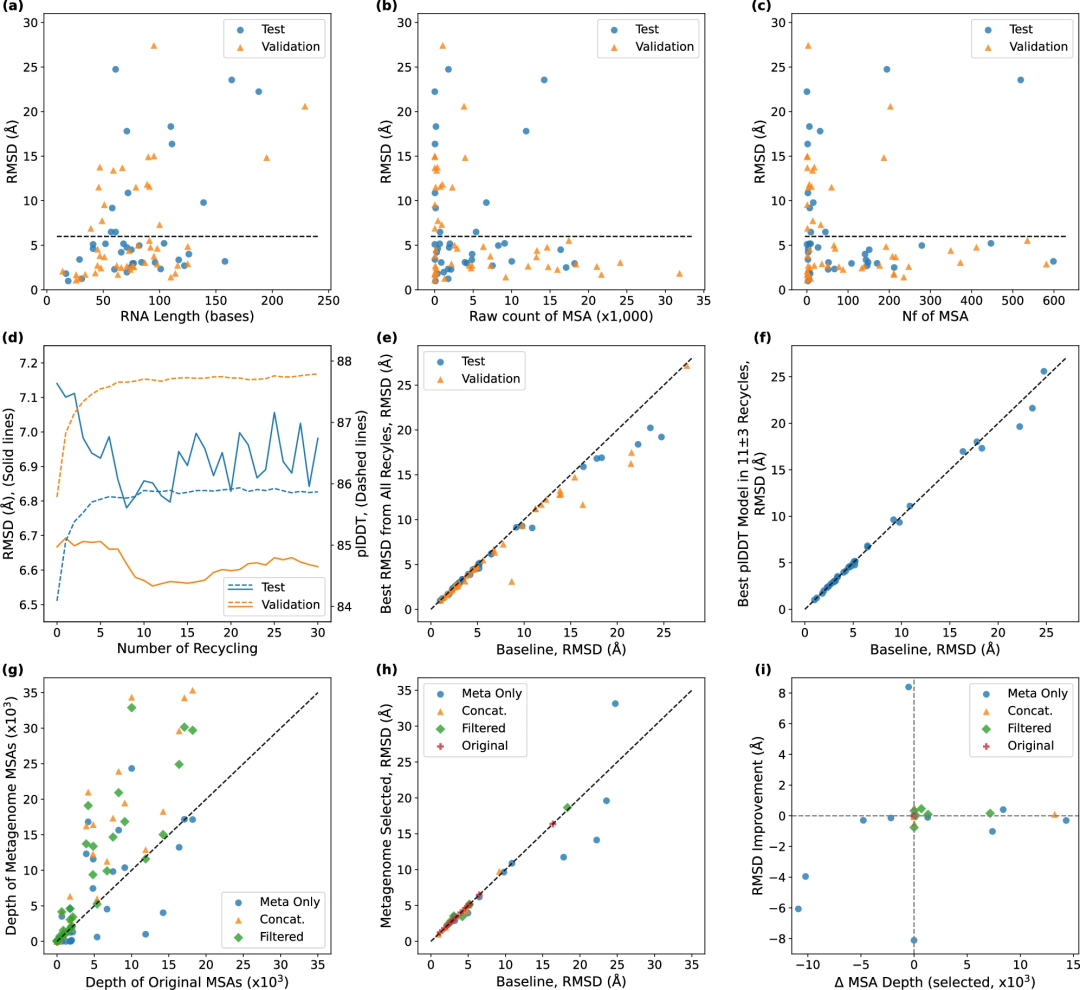
图示:目标长度、MSA 深度、回收和宏基因组 MSA 对建模准确性的影响。(来源:论文)为了增加输入 MSA 的深度,科学家加入了宏基因组序列;事实证明,利用宏基因组序列作为输入 MSA 并优化循环次数可提高 NuFold 的预测性能。在训练过程中,他们采用了动态采样策略,在易目标和难目标之间保持平衡。
此外,团队还测试了几个较小的网络,因为他们拥有的训练数据比原始 AF2 中使用的蛋白质数据集要小。NuFold 对大多数测试目标实现了 5 Å 或更低的全原子 RMSD,几乎完美地构建了构象,柔性末端和环状区域除外。
未来
虽然 NuFold的表现明显优于基于能量最小化的方法,但表现略差于近期发布的一些基于深度学习的方法。
一个原因是,训练数据量不足以支持这种完全原子级详细模型。训练数据不足对于任何 RNA 结构预测方法来说都是一个挑战,但对于 NuFold 来说,这可能更为关键,因为它直接从深度神经网络建模完整的原子结构。
为了解决这个问题,结合不同的数据模式非常重要,例如提供 RNA 二级结构洞察的实验数据,如 DMS-MaPseq 和 SHAPE-MaP。
关于未来的工作,由于 RNA 的构象会受到与其他分子相互作用的影响,因此多链、RNA 和蛋白质的结合是 NuFold 的一个扩展。除了 RNA 和蛋白质之外,使用小化合物建模也是一个重要的扩展,因为 RNA 是药物发现的新兴目标。
源代码:https://github.com/kiharalab/nufold/
论文链接:https://www.nature.com/articles/s41467-025-56261-7
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










