将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

编辑 | KX
预测蛋白质构象变化是计算生物学和人工智能领域的一大挑战。主流的 AlphaFold 等算法可以高通量预测蛋白质的静态结构,但对蛋白质构象变化预测却束手无策。
为了解决这个问题,中国科学技术大学和上海科技大学的研究人员,提出了一种新颖的深度学习策略,即利用高通量生物物理采样来规避与蛋白质构象转变相关的数据匮乏。
研究人员将分子动力学模拟与增强采样方法相结合,创建了一个大规模数据库。研究模拟了 2635 种已知两种稳定状态的蛋白质的构象变化,并收集了每条转变途径的结构信息。利用这个数据库,开发了一个能够预测给定蛋白质转变途径的通用深度学习模型。
该模型对不同序列长度(从 44 到 704 个氨基酸)的蛋白质表现出普遍的稳健性,并适应不同类型的构象变化。在几个系统中,预测和实验数据结果高度一致,并成功地应用该模型来识别一个重要生物系统——人类 β-心脏肌球蛋白中的一种新的变构调控。
这些结果证明了该模型在揭示蛋白质构象变化本质方面的有效性。
相关研究以「Exploring Protein Conformational Changes Using a Large-Scale Biophysical Sampling Augmented Deep Learning Strategy」为题,发表在《Advanced Science》上。

论文链接:https://doi.org/10.1002/advs.202400884当前局限于蛋白质静态结构预测
AlphaFold 和 RoseTTAFold 等致力于预测蛋白质的静态结构,但并未解决大多数蛋白质在发挥其生物学作用时所经历的动态构象变化。
这些转变对于理解从酶活性到信号转导的广泛生物过程至关重要。然而,缺乏中间状态的结构数据阻碍了对这些转变的预测。此外,现有模型的过渡态自由能垒较高,使得准确预测更具挑战性。
现有的描述蛋白质构象转变的模型包括基于弹性网络的正常模式分析,以及将弹性网络与分子动力学模拟相结合的混合模型。这些方法适用于相当简单的构象运动,但无法解释在较大的蛋白质中发现的复杂和巨大的变化。
最近,深度学习方法将蛋白质结构映射到低维潜在空间。然而,这些模型依赖于两种状态之间的线性路径,这不适用于复杂的非线性转换,例如倍数切换。更重要的是,高数据需求和低数据效率,以及阻碍实时可扩展应用的计算成本,使得这些方法本身无法令人满意。
模拟 2635 种蛋白质的构象变化
受深度学习在预测静态蛋白质结构方面的成功启发,研究人员现在正在积极探索其他旨在预测蛋白质构象变化的深度学习算法。其主要挑战在于表征不同构象转变的训练数据有限。
在此,中科大团队采取了更直接的方法来弥补数据短缺的问题。通过结合分子动力学模拟和增强采样方法,广泛模拟了 2635 种蛋白质的构象变化,其中包含两种实验确定的状态,即单状态 (SS) 和多状态 (MS) ,并直接获得了组成转换途径的结构。
SS 和 MS 蛋白质数据集的创建涉及对蛋白质数据库 (PDB) 中的结构进行全面搜索和比较。
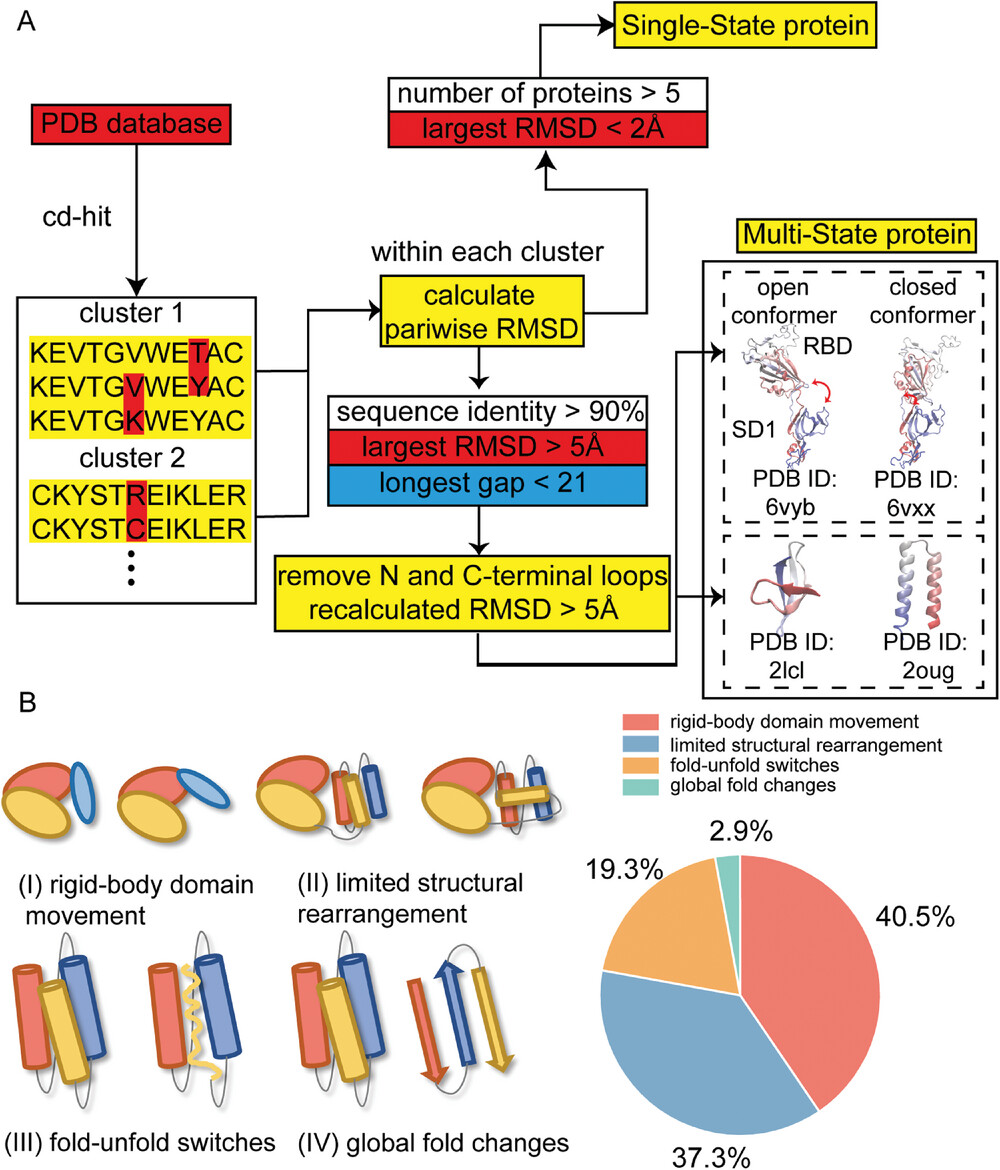
图示:创建单状态 (SS) 和多状态 (MS) 蛋白质数据集。A) 数据集创建的工作流程。B) MS 数据集中四个类别的统计分析。(来源:论文)SS 数据集共包含 3454 个条目,包括在实验研究中观察到的具有高度收敛构象的蛋白质。相比之下,MS 数据集包含 2635 种蛋白质(每种蛋白质在 PDB 中都有两种结构),这些蛋白质表现出显著的构象变化,定义为均方根偏差 (RMSD) >5 Å。
该数据集可进一步分为四个子类:刚体结构域移动(rigid-body domain movement)、有限的结构重排(limited structural rearrangement)、折叠-展开切换(fold-unfold switches)、全局折叠变化(global fold changes )。
高精度预测蛋白转变途径的通用 AI 模型
该数据集使用一种称为 PATHpre 的整体深度学习模型,可以高精度地预测导致构象转变的结构途径。
PATHpre 中 HESpre 模块的核心创新在于预测转变途径中高能状态的性能。所提出的模型对不同构象的蛋白质具有极大的通用性。这意味着它对复杂系统中的动力学行为建模做出了巨大贡献,在方法级别应用了可扩展性和数据效率。
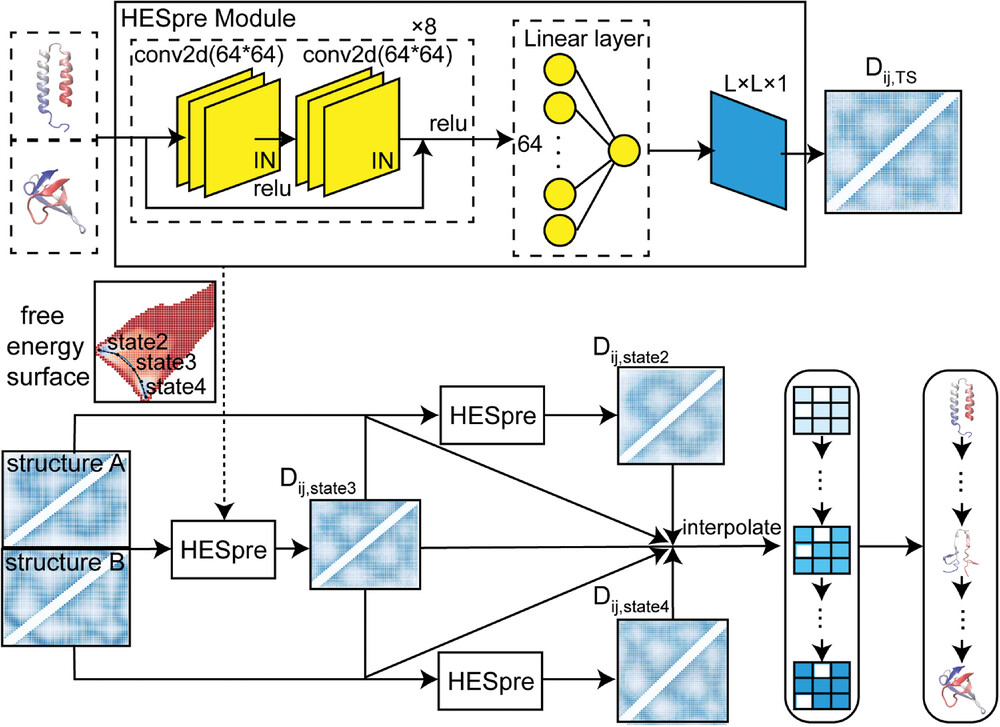
图示:用于预测多构象蛋白质过渡途径的神经网络架构。(来源:论文)在 PATHpre 方法中,通过卷积神经网络预测应用两种构象状态系统中的距离矩阵,以获得这些构象状态之间的高能状态。核心模块名为 HESpre,目的是预测高能状态下的结构信息。然后迭代使用该模块来预测整个途径。
研究对各种蛋白质进行了交叉验证,该模型在所有步骤中都实现了强大的 Pearson 相关性和低平均绝对误差;因此,它在结构类别中非常通用。
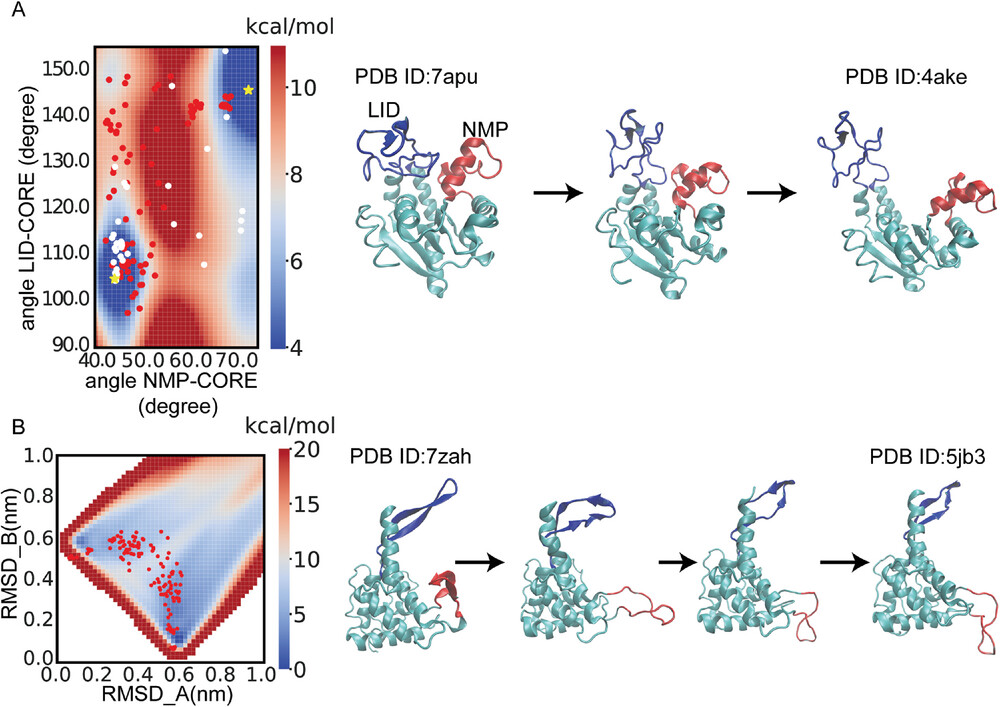
图示:应用 PATHpre 研究两种蛋白质的构象变化:A) 腺苷酸激酶和 B) 30S 核糖体蛋白 S7。(来源:论文)PATHpre 通过与一系列蛋白质上存在的实验和模拟数据显示出强相关性,在蛋白质转变途径非常高的预测中很准确。评估还表明,PATHpre 可以稳健地捕获从简单到复杂的构象变化,并且它与不同长度的序列以及结构复杂性保持一致。
重要的是,它通过匹配实验自由能景观,准确预测了单个蛋白质(如腺苷酸激酶和 30S 核糖体蛋白 S7)的转变途径,并且在具有挑战性的条件下,比传统的混合方法表现更好。PATHpre 的预测与已知结构一致,其对折叠转变蛋白质中精细中间状态的映射证实了其广泛的适用性和可靠性,可以捕获广泛的蛋白质构象转变。
蛋白质构象变化新见解
与其它几种同类蛋白质构象变化深度学习模型相比。PATHpre 模型具有以下特点:
首先,PATHpre 使用来自模拟的转变数据进行训练,消除了转变路径在潜在空间中呈线性的假设。
其次,模型是适用于所有蛋白质的通用模型,无需针对每种蛋白质进行自定义再训练。
第三,模型专注于结构明确的蛋白质的大构象变化(RMSD > 5Å),而不是内在无序的蛋白质或局部变化相对较小的蛋白质,这在药物设计中很重要。
该研究标志着 AI 驱动的蛋白质建模取得了重大进展,为预测蛋白质构象变化提供了一种数据高效且可扩展的方法。该模型将进一步提高科学家对蛋白质构象变化的理解。
数据库和源代码地址:https://github.com/qwang897/PATHpre
参考内容:https://www.marktechpost.com/2024/11/03/a-study-on-protein-conformational-changes-using-a-large-scale-biophysical-sampling-augmented-deep-learning-strategy/
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










