
近年来,理论计算与机器学习,越来越受顶刊青睐!“实验+计算”的模式已成为顶刊标配,机器学习亦逐渐成为一种强有力的研究工具!
经过1000余天的打磨和改进,华算科技陆续推出Materials Studio计算、LAMMPS计算、VASP计算、机器学习与材料等培训,备受好评,往期学员已陆续在Nature子刊、Angew.、JACS、AM等顶级期刊发表文章!

线上课程,不限时间、不限地点、随报随学、无限次回看。
课程群永不解散,随时提问,及时解答。提供超算练习账号,边学边练。
报名方式:添加下方微信好友,或者联系手机130-0542-7160。
👇👇 扫描二维码,立即报名 👇👇

👆👆 电话:130-0542-7160 👆👆
带你零基础入门DFT计算/机器学习,提升论文档次,发顶刊快人一步!
学员已发表Nature、Nature Energy、Nature Materials、Angew..、JACS、AM、AEM等期刊。

专题20:VASP晶体材料计算培训:晶体结构、电子、弹性、光学、磁性、电池、催化性质计算
专题21:VASP低维材料计算培训:二维/一维/异质结的结构、缺陷、电子、电池、吸附与催化性质计算专为零基础学员设计,采用CASTEP/DMol3模块授课,沿着理论讲解、模型搭建、性质计算、结果分析层层递进。
全面覆盖从三维晶体到二维表面(包括吸附、催化反应中的中间态结构)、二维材料、异质结构、固液界面,再到一维纳米管与零维团簇,从分子到聚合物,再到复杂COF结构与超分子的构建。既包含纯相,又涵盖缺陷/掺杂模型与吸附构型。计算部分涵盖能带、态密度、PDOS、电荷布居、差分电荷密度、COHP、功函数、光学/弹性性质、声子谱与态密度、热力学性质、表面能、吸附能、过渡态,涉及体系包括二维材料、体块材料与表面。基于Materials Studio计算平台CASTEP, DMol3模块设计,直击催化科研热点,包括了催化理论、建模、热力学、动力学、吸附性质分析等内容。包括学员们非常关心的CO2RR/OER/HER自由能台阶图、火山理论、d带中心、过渡态搜索/确认/优化/频率验证、吸附与活化、COHP、掺杂改性、带隙/带边位置调整、光生载流子分离、异质结构能带电位匹配、层间差分电荷密度等科研热点。反应热力学+动力学性质:Li+/Na+脱出/嵌入过程中电位变化曲线、克容量、离子扩散路径、过渡态能垒。
电子结构性质:磁性、晶体场劈裂、姜泰勒畸变、DFT+U、PDOS、COHP、电荷布居、差分电荷密度分析;Li与(复合)电极材料的结合、阴离子氧化还原。课程主要知识点:分子动力学+AIMD、电解液建模、固态电解质、RDF+配位数、SSIPs+CIPs+AGGs、MSD+离子电导率、离子迁移孔道+势垒、溶剂化效应+溶剂化能、HOMO/LUMO+静电势。课程主要涵盖钙钛矿太阳能电池、光吸收、介电函数、介电常数、有效质量、载流子迁移率、金属半导体异质结构、Janus异质结构、内建电场、整流效应、气敏半导体等。课程主要涵盖量子化学理论、分子建模、轨道、电荷分布、静电势、振动、自由能、解离能、过渡态、IRC、势能面扫描、光谱、激发态、核磁共振、弱相互作用、溶剂化、pKa等计算。高熵合金、多晶孪晶、聚合物、粗粒化、拉伸压缩、压痕冲击、结晶、传热扩散、交联反应、玻璃化转变温度、径向分布函数(RDF)、概率分布函数(PDF)、 扩散系数、均方位移(MSD)、位错分析、表面张力、氢键寿命课程内容涉及晶体与二维材料材料的建模、弹性、电子(电荷态、态密度、轨道杂化、能带、功函数)、光学、磁性、吸附、催化性质计算,帮助大家快速理解模型构建与VASP输入文件中各个参数的意义,提高学习效率,完成DFT计算入门。课程具体涉及:理论知识、二维/异质结构建模、能带、功函数、静电势、吸附位点、吸附能、结合能、虚频消除、态密度、d带中心、过渡态、CO2RR、OER/ORR、电位、容量、迁移势垒。课程内容涉及晶体表面/二维结构/一维结构的结构性质,功函数、d带中心、吸附能、差分电荷密度等电子性质,HER、OER/ORR、NRR、CO2RR等催化反应的自由能与过渡态计算,CO、CO2、CH4、C6H6等分子的吸附与分解过程。单原子、双原子催化剂是目前电催化领域研究的最多的催化剂材料,他们具有原子利用率高、活性高、性能精确可调的特点,能够有效地催化各种化学反应,包括HER、OER、CO2RR、NO3RR等,在能源、化工等领域起着重要作用。DFT计算能够深入研究单原子催化剂结构稳定性、电子结构、对反应中间体的吸附能力、以及对反应热力学和动力学势垒的影响,已经被广泛应用于催化剂设计、筛选、性能分析等方面。本次课程由华算科技朱老师主讲单原子催化计算,课程具体涉及催化剂模型,电子性质,HER、OER/ORR、CO2RR、NO3RR等反应的自由能与过渡态计算。催化剂模型:表面结构、单原子与双原子、纳米颗粒、合金、高熵合金、异质结构电子性质:态密度、d带中心、差分电荷密度、静电势、功函数HER:反应自由能,PH值影响,中间体H2O、OH、H构型,过渡态,势能面,虚频消除,台阶图
OER/ORR:反应路径,中间体OH、O、OOH构型,线性约束关系,电位影响,台阶图CO2RR:CO路径、HCOOH路径、CH3OH路径、CH4路径、台阶图NO3RR:反应路径,NO、NH2、ONH等中间体构型、台阶图NRR:distal路径、alternating路径、enzymatic路径、台阶图DFT计算在催化性质研究方面具有明显的优势,能够准确计算反应势垒、反应路径、中间体构型、电子结构、电荷转移等重要物理化学参量。VASP是目前最流行的DFT计算软件,具有功能强大、计算效率高等优势。本次课程由华算科技朱老师主讲非金属催化计算,课程具体涉及热门催化剂对HER、OER/ORR、CO2RR、NO3RR、NRR反应的催化性能。材料的磁性受到原子磁矩大小和排列方式的影响,宏观上表现为顺磁、铁磁、反铁磁、亚铁磁等特性。在居里温度或奈尔温度下材料的磁性能发生转化,即从铁磁/反铁磁性转变为顺磁性。材料的磁性涉及较复杂的物理理论,包括自旋轨道耦合、量子霍尔效应、反常量子霍尔效应等,对初学者来说有较大的难度。本次课程通过VASP软件、Python软件、蒙特卡洛方法、紧束缚模型等工具给大家介绍材料磁性的计算理论与计算方法,具体涉及VASP输入输出文件,铁磁/反铁磁/亚铁磁构型、磁各向异性、晶体场、磁相变温度、四方/三方/六角格子的紧束缚模型求解、反常量子霍尔效应、贝里曲率和陈数。本次课程将运用VASP软件计算钙钛矿半导体的结构、电子、吸附、缺陷性质,二维半导体的结构与电子性质。课程内容将根据学员反馈不断更新。运用VASP软件计算三维和二维半导体材料的能带、态密度、缺陷形成能、扩散势垒、吸附能、电荷转移性质。内容涉及建构建模、电荷密度、态密度、能带结构、缺陷形成能、缺陷转变能级、缺陷扩散路径与势垒、吸附能、结合能、差分电荷密度计算。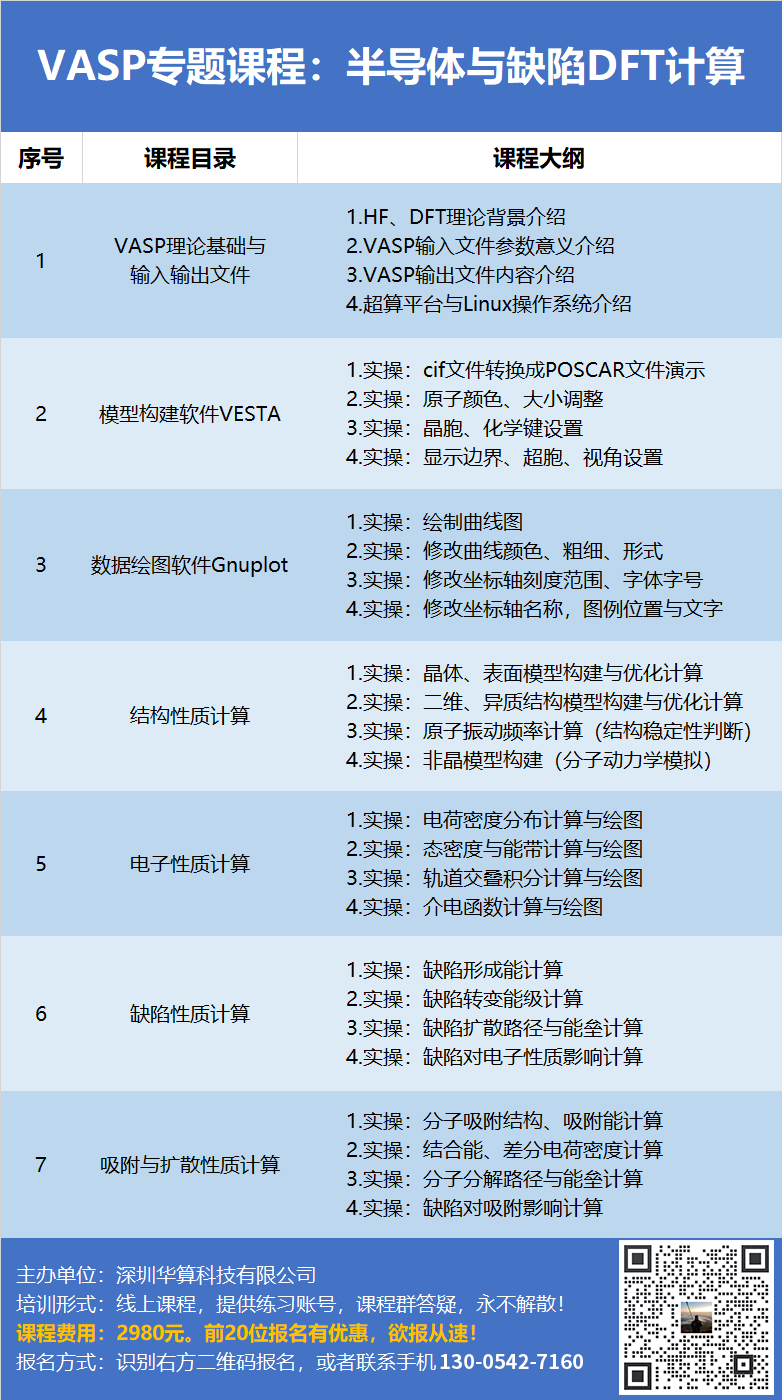
课程涉及电池中的电化学反应类型(插入、合金、转化)的判断,正负极材料(三维、二维、二维异质结构)的掺杂、离子容量、离子扩散能垒计算,离子与正负极材料的相互作用机理计算,燃料电池所涉及的H2氧化与O2还原过程决速步、过电位的计算。
DFT计算能够深入研究燃料电池所涉及的催化剂对电化学反应的催化过程。例如,在Pt、Ru等贵金属催化下氢氧燃料电池阳极和阴极所发生HOR和ORR的反应速率明显得到提升。本次课程由华算科技朱老师主讲燃料电池计算,课程具体涉及催化剂模型、电子结构,反应中间体吸附、反应动力学等等。DFT计算能够深入研究Li-S电池所涉及的多硫化锂吸附与转化过程。单原子、双原子、金属等类型催化剂被证明具有较好的催化性能。DFT计算软件VASP在多硫化锂吸附与转化计算方面具有较强的优势,能够准确计算反应自由能、热力学与动力学势垒、吸附构型、电荷转移等重要性质。本次课程由华算科技朱老师主讲Li-S电池计算,课程具体涉及催化剂模型,电子结构,多硫化锂吸附构型、转化反应自由能、分解反应动力学。
通过VASP软件的晶体结构、电子、弹性、磁学性质,二维材料的结构、吸附、扩散性质的计算练习,帮助大家快速理解模型构建与VASP输入文件中各个参数的意义,提高学习效率,完成DFT计算入门。
本次课程将介绍二维材料、一维材料、二维材料异质结构的结构、缺陷、电子、电池、吸附与催化性质。课程针对编程零基础学员,并包含实验与计算中常见的科研数据处理案例。同学们学习完成后可直接将Python应用于自己的研究数据后处理之中,适合有数据分析需求的实验课题组/计算课题组同学。课程面向Python零基础,对机器学习感兴趣,想在自己的研究方向使用机器学习的化学、材料学相关工作者。通过本次课程,大家可以学会当下最流行的Python语言,学会抓取数据库,能使用机器学习基本算法,并会用于机器学习解决化学与材料学的实验数据处理、材料筛选与性质预测等问题,能够重现机器学习的文献案例。课程包括:机器学习概念,决策树/贝叶斯/支持向量机/神经网络/随机森林,回归/分类/聚类算法,模型评价/性能度量,小分子/MOF/d带中心/合金催化/单原子催化/钢铁强度/CO2RR/钙钛矿/电极涂层/STM图像,数据库/高通量筛选/Materials Project。课程包括:线性模型/模型评价/约束项/神经网络/分类算法/回归算法/集成学习/XGBoost/描述符/预处理课程须知:本次课程报名需有一定的Python代码基础,零基础同学请选择《Python数据分析》课程。本次课程主要基于Python语言与TensorFlow/Keras框架,学习神经网络的构建、训练与使用,课程中使用的绝大部分案例均源于化学/材料文献中真实案例,同学们学习完成后可直接将其用于自己的研究当中。课程设计目的为学习完成之后,能自己构建常用的神经网络,进行各种技巧处理,并进行应用。课程须知:本次课程报名需有一定的Python代码基础和机器学习基础,零基础同学请选择《Python数据分析》《机器学习与材料/化学》课程。主办单位:深圳华算科技有限公司(拥有VASP、Materials Studio、Gaussian、LAMMPS商业版权)
培训形式:课程已上架小鹅通,可无限次回看。课程群永不解散,随时提问,及时解答。课程费用:咨询华算科技-邓庆烽,提供增值税普通发票及邀请函。同时报名两个课程可优惠1600元,更多优惠咨询可咨询华算科技-邓庆烽。报名方式:添加下方微信好友报名,或者联系手机130-0542-7160 。杨老师:华算科技全职技术资深专家,深圳市孔雀计划海外高层次人才。曾就职于德国马克思普朗克研究所,日本WPI研究所,并曾在芬兰阿尔托大学进行长期访问,作为PI主持欧盟、日本科研项目6项。拥有10年以上Materials Studio软件使用经验,主要从事固态相变的第一性原理研究、电化学固液界面的AIMD研究与超分子化学中的分子动力学模拟。朱老师:华算科技资深技术,同济大学本科直接攻读博士学位(4年),海外3年以上博后经历,发表高质量独立一作SCI论文30篇,回国后被授予深圳市海外高层次人才。拥有14年VASP重度使用经验,成功讲授100+场VASP计算培训和超过10万人的学习理论计算公开课。黄老师:华算科技全职技术专家,武汉大学本科,北京大学博士,新加坡国立大学访问学者。目前已发表SCI文章共20篇,其中第一作者文章5篇,单篇最高影响因子>40。从事理论计算与实验化学研究工作11年,擅长使用机器学习进行化学理论的研究及实验数据的处理,曾获华中地区数学建模邀请赛三等奖,北京大学游戏AI对抗全国邀请赛第四名等相关奖项。LAMMPS王老师:
华算科技资深技术专家,六年以上LAMMPS软件重度使用经验。发表SCI共10篇,并参与编写英文书籍《Fundamentals of Multiscale Modeling of Structural Materials》。
主要研究领域为:利用分子动力学模拟方法,①研究金属(梯度和中、高熵合金)及其纳米复合材料的力学性能;②开发高分子和石墨烯的粗
粒化模型,并研究高分子及其纳米复合材料的结构、动力学和力学性能。
VASP磁性-张老师:高校教师,博士毕业南京大学,凝聚态物理专业。从事自旋电子学、铁电、压电、多铁、谷电子学、微磁学模拟等领域的理论研究。擅长使用VASP做数值计算,尤其在磁性材料方面,具有丰富的数值计算和模型计算经验。在拓扑绝缘体、磁性拓扑半金属、磁性谷电子学、压电材料等领域发表多篇学术期刊。在Phys. Rev. B、Appl. Phys. Lett.、 Appl. Phys. Rev.、 Adv. Func. Mater.、Nanoscale、PCCP、J. Phys. Conden. Matter等期刊发表学论文20多篇。








