
原子尺度模拟已成为提供实验观察现象微观解释的宝贵计算工具,但传统的物理模型势函数在计算成本、准确性和广泛适用性之间存在权衡,尤其在模拟反应化学时更为明显。传统的力场和量子力学方法在计算成本和适用范围上存在限制,而反应力场则需要根据预定的反应进行重新参数化,量子力学方法的计算成本对许多反应性分子动力学研究来说是禁止的。
近日,来自美国卡内基梅隆大学的Shuhao Zhang,洛斯阿拉莫斯国家实验室的Nicholas Lubbers, Richard A. Messerly & Justin S. Smith等人在 Nature Chemistry 期刊发表了题为“Exploring the frontiers of condensed-phaseb chemistry with a general reactive machine learning potential”的论文,本文讨论了开发一种用于研究凝聚相系统中反应化学的通用反应机器学习势能(ANI-1xnr)的方法。该方法涉及自动采样凝聚相反应,以创建适用于C、H、N和O元素的高度通用的反应MLIP。该方法利用主动学习来生成用于训练ML模型的多样且相关的数据集。MLIP直接训练到凝聚相QM反应数据,以确保对反应MD模拟的可靠性。研究包括碳成核、石墨烯形成、生物燃料添加剂、甲烷燃烧以及甘氨酸的自发形成等应用。

图1 纳米反应器主动学习工作流程和所考虑的具体应用的总结 ©2024 Springer Nature
AL 循环是一种用于开发 MLIP 的自动化、迭代且高效的方法。AL 生成一个训练数据集,其中仅包含基于 MLIP 集合识别的高不确定性结构的量子计算。使用 NR 模拟对与凝聚相反应化学相关的结构进行采样。初始系统由由元素 C、H、N 和 O 组成的小分子的随机配置构建。使用当前的 MLIP 进行动态模拟,在温度和体积的极端波动下诱导化学反应。为了测试所得模型的通用性,最终的 MLIP 随后应用于训练期间未直接针对的几个案例研究。

图2 使用纳米反应器主动学习分析本工作中生成的数据集 (ANI-1xnr) ©2024 Springer Nature
在评估 ANI-1xnr 模型在不同案例研究中的性能之前,作者评估了 ANI-1xnr 数据集的多样性和完整性。图 2 通过将元素 H、C、N和 O的相似局部原子环境聚集在一起,提供了高维数据集的二维可视化。图 2a-d 比较了 ANI-1xnr 数据集和非反应性、接近平衡的真空分子 AL 数据集 (ANI-1x)。显然,ANI-1xnr 数据集不仅有效地涵盖了整个 ANI-1x 数据集,而且还大大超出了 ANI-1x 覆盖的局部原子环境空间。更重要的是,ANI-1xnr 数据集提供了 ANI-1x 数据集中许多簇之间的路径。这些途径可能对应于低维表示中的反应。此外,图 2e 提供了 ANI-1xnr 训练数据集中识别的 1000 多个独特分子的精选示例。由于 NR 采样模拟仅使用小分子进行初始化,因此 NR-AL 程序自动发现了数百条导致这些不同分子结构的反应途径。
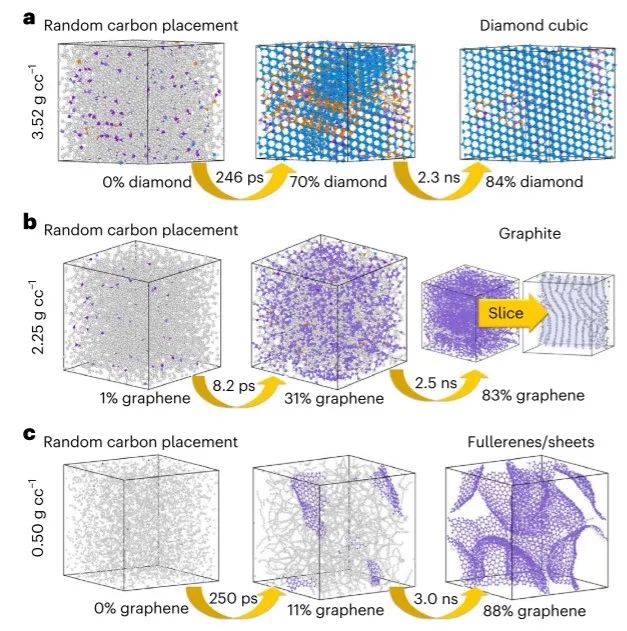
图3 ANI-1xnr 的碳固相成核模拟结果 ©2024 Springer Nature
图 3 总结了每次模拟的结果。对于每个高密度、中密度和低密度碳模拟,ANI-1xnr 生成相应密度的预期碳结构。具体来说,对于密度最高的体系,246 ps后,金刚石、石墨烯和六方金刚石相共存,其中模拟盒中70%的碳原子形成金刚石立方晶体结构。又过了 2.3 ns,高密度体系在金刚石立方晶体结构中含有 86% 的碳原子,石墨烯和六方金刚石位点很少。在中密度体系中,8.2 ps 后,31% 的原子快速形成石墨烯,再过 2.3 ns 后,体系中含有 83% 的石墨烯。石墨烯片倾向于形成堆叠且更有序的类石墨结构。低密度系统在 250 ps 后形成碳原子链,其中 11% 的原子形成石墨烯片。再过 3 ns 后,系统包含 88% 的石墨烯片中形成的原子。
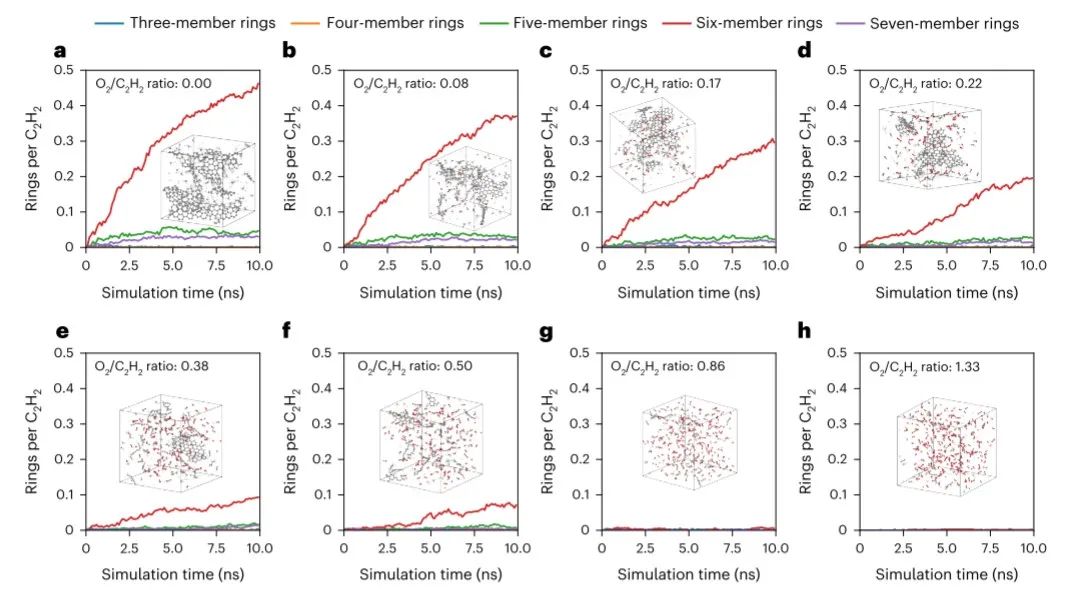
图4 氧对 ANI-1xnr 石墨烯成环模拟结果的影响 ©2024 Springer Nature
图 4 显示了在八种不同的 O2/C2H2 比率下,三元、四元、五元、六元和七元环的形成随模拟时间的变化情况。它展示了随着 O2/C2H2 比率的变化,形成的环数量随时间的变化情况。该图直观地展示了系统中氧气相对于乙炔的浓度如何影响不同大小环的形成。与 Lei 等人的模拟相比,ANI-1xnr 与实验数据的一致性有所提高,这主要是由于 ANI-1xnr 的模拟时间尺度更长、系统规模更大、计算成本更低。
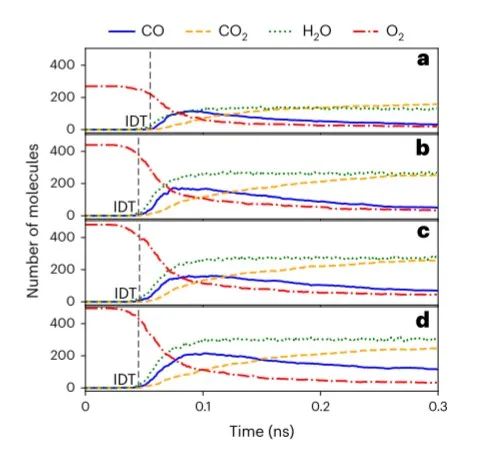
图5 ANI-1xnr 生物燃料添加剂模拟结果 ©2024 Springer Nature
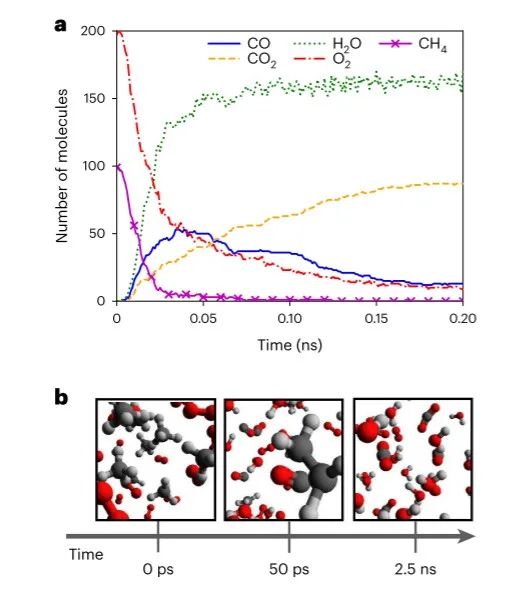
图6 ANI-1xnr 的甲烷燃烧模拟结果 ©2024 Springer Nature
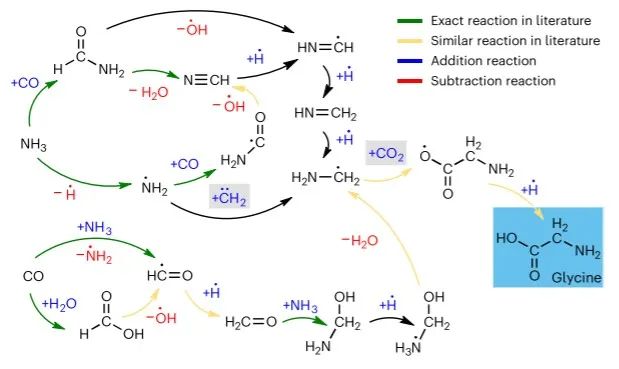
图7 ANI-1xnr 的米勒实验模拟结果 ©2024 Springer Nature
为了进一步证明了 ANI-1xnr 乙醇模拟结果的正确性,我们重现了 Chen 等人模拟的四个系统,实验结果表明乙醇可以在相对较高的压力下加速燃料点燃,这与模拟结果一致。这表明 ANI-1xnr 电位准确地捕捉到了乙醇作为燃料添加剂在高压条件下的行为。
图 6a 显示 ANI-1xnr 潜力产生的主要产品和物种概况与 Zeng 等人的非常相似。然而,与 Zeng 等人的 CH4 和 O2 消耗速率相比,ANI-1xnr 预测总体反应速率大约快 40 倍。他们的系统需要 0.5 ns 的模拟时间来消耗一半的初始 CH4 ,而作者的系统只需要 0.012 ns。
图 7 显示了从初始反应物开始形成甘氨酸的 ANI-1xnr 反应机制。在我们的米勒模拟过程中,甘氨酸形成了三次,并持续了大约 225 fs、375 fs 和 913 fs。考虑到该模拟的相对较高的温度,预计甘氨酸的解离时间小于 1 ps。形成甘氨酸的最后一步是向 C2H4NO2 进行氢加成,类似于 Saitta 和 Saija 的机制。然而,在我们的机制中,氢加成发生在氧原子上,而不是像 Saitta 和 Saija 机制中那样发生在 α-碳上。
总之,ANI-1xnr 潜在的未来改进包括利用低密度或真空反应数据增强凝聚相数据集,将数据集扩展到更多元素,以及研究更高精度量子力学方法的前景,以获得更好的反应障碍。此外,未来的工作还可以探索更先进的机器学习训练范式,如迁移学习、元学习和终身学习。关于模型的形式,未来的工作可以研究如何结合最新的发展,如明确的长程项和电荷依赖性神经网络电位。
至于在训练领域之外应用 ANI-1xnr 时的注意事项,潜在用户应了解 ANI-1xnr 只在特定密度范围的凝聚相模拟中进行过训练。因此,不建议将其用于空泡计算或密度差异较大的系统。此外,用户在将 ANI-1xnr 应用于涉及气相中从单重态到三重态电子状态变化的系统时应谨慎,因为 ANI-1xnr 并未针对此类情况进行明确的训练。
原文详情:
Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential. Nat. Chem. (2024).
DOI: 10.1038/s41557-023-01427-3
本文由尼古拉斯供稿。
cailiaorenVIP











