将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


编辑 | 萝卜皮
与 DNA 和 RNA 不同,蛋白质缺乏准确和高通量的测序方法,这阻碍了蛋白质组学在序列未知的应用中的实用性,包括变体调用、新表位鉴定和宏蛋白质组学。
德国慕尼黑工业大学(Technische Universität München,TUM)的研究人员推出了 Spectralis,一种用于串联质谱分析的从头肽测序方法。Spectralis 利用了多项创新,包括连接按氨基酸质量间隔的光谱峰的卷积神经网络层、提出碎片离子系列分类作为从头肽测序的关键任务,以及肽谱置信度评分。
对于数据库搜索提供的真实光谱,Spectralis 的灵敏度超过 40%,精度高达 90%,几乎是当前 SOTA 灵敏度的两倍。对未识别光谱的应用证实了其优越性,并展示了其对变异调用的适用性。
该研究以「Deep learning-driven fragment ion series classification enables highly precise and sensitive de novo peptide sequencing」为题,于 2024 年 1 月 2 日发布在《Nature Communications》。

液相色谱串联质谱法是高通量鉴定蛋白质的首选方法。为此,蛋白质首先被消化成肽,其质荷比(m/z)在第一质谱中确定。接下来,选定的肽沿着其主链键断裂,生成一系列肽片段,其 m/z 比可以在第二个质谱中识别。原则上,该光谱允许通过读出相同离子系列的连续峰之间的 m/z 差异来重建肽序列。
在实践中,由于缺少峰、污染峰,并且峰的离子系列事先未知,因此这项任务非常困难。当将实验光谱与有限的一组可能的肽(通常是所研究的生物体的计算机消化的蛋白质组)的预期光谱进行比较时,肽的鉴定将得到极大的促进。
这种策略需要预先计算可能的肽的数据库,称为数据库搜索。绝大多数蛋白质组学研究依赖于数据库搜索,尽管数据库搜索在设计上不允许识别新的或意想不到的肽。这阻碍了蛋白质组学在肽序列事先未知的应用中的有效使用。这涉及新表位鉴定、抗体测序、病原体监测、微生物群落研究和古生物学。因此,非常需要高效的从头肽测序算法,其目的是直接从光谱中识别肽,而不依赖于任何数据库。
大多数从头肽测序算法都采用组合优化方法,其中搜索最适合光谱的肽。各种肽谱匹配(PSM)分数,即评估候选肽与给定谱的对应程度的分数,与包括动态编程和遗传算法在内的组合优化技术相结合,已被用来识别最适合的肽。然而,缺失峰和污染峰严重限制了这些算法的准确性。
与这项工作并行的是,科学家们利用深度学习在前向问题上取得了重大进展,即预测给定肽序列的光谱。虽然这些算法不能预测污染峰,但它们可以预测给定肽的峰强度和缺失峰。因此,可以利用他们的预测为 de novo 肽测序算法(如算法 pNovo3 中的算法)开发更具辨别力的 PSM 评分函数。
作为对这些基于组合优化的算法的补充,最近提出了直接预测光谱中肽序列的神经网络。这包括 DeepNovo、PointNovo 和 Casanovo。尽管如此,现有的从头肽测序方法的性能仍然有限,特别是在高精度范围内的灵敏度较差。需要进一步改进方法,以增加串联质谱实验中高度可信的肽序列鉴定的数量。
慕尼黑工业大学的研究团队推出了 Spectralis,一种结合了多种算法创新的方法,用于从头肽测序。Spectralis 以该领域的既定概念为基础,例如基于碎片模式的频谱图和 PSM 评分函数,并利用深度学习模型进行频谱预测 Prosit。
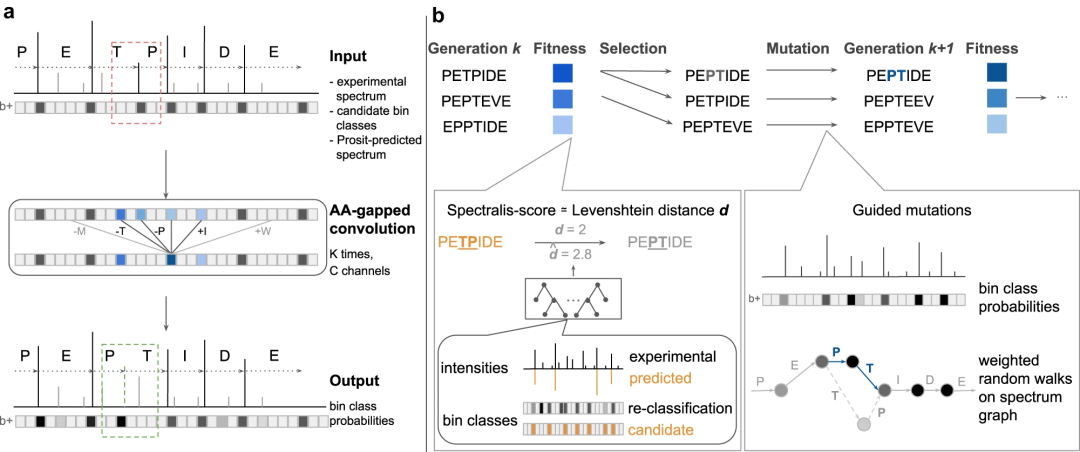
图:Spectralis 的 Bin 重新分类和概述。(来源:论文)Spectralis 的核心由监督学习任务组成,该团队称之为 bin 重分类。该研究表明,预测的 bin 类别可以提高 PSM 的评分。使用 Levenshtein 距离估计作为 PSM 分数,研究人员证明了对现有从头肽测序方法预测的肽进行重新评分可以在 90% 的精度下将召回率提高近两倍。此外,该团队利用这些建模创新设计了一种进化算法,从而提高了整体召回率。
这里的分数不会提高整体召回率,因为它不会修改预测的肽。然而,在实践中,对于实现正确预测的肽和错误预测的肽之间更好分离是非常有利的。评分函数可以用作独立方法,排名的计算成本很小。它还允许使用单一程序对多种从头肽测序方法中预测的肽进行比较和整合。
引导突变在改善不正确的候选肽方面显示出有希望的结果。然而,利用引导突变的进化算法比单独重新评分产生了适度的改进。
尽管如此,研究人员发现它可以生成几个与正确肽的编辑距离非常小的看似合理的候选肽。考虑每个光谱的两个或多个高置信度预测对于识别大部分肽(但不是全部)感兴趣的应用可能很有用,例如在光谱不明确的情况下。然而,对于高精度用例,应优先将他们的分数应用于现有从头测序工具提出的候选肽。
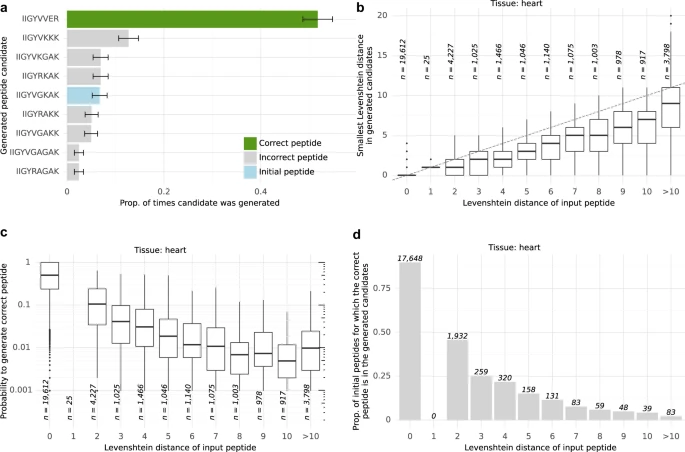
图:引导突变性能。(来源:论文)
用于导出引导肽突变的图,让人想起广泛使用在实验光谱峰上定义的光谱图。与基于峰的谱图相比,该团队的图表示的一个限制是他们以 1 道尔顿分辨率进行操作。尽管 1 道尔顿大约相当于质子或中子的质量,但质谱仪允许以更高分辨率进行测量,这在原则上是可以利用的。
不过,1 道尔顿分辨率并不是该方法的概念限制。可以以更长的运行时间为代价获得更高的分辨率。基于 bin 的图而不是基于峰值的图的优点是,图的节点不依赖于实验峰值的存在,而仅取决于 bin 重新分类的输出。因此,这有利于生成连接由单个氨基酸质量间隔的节点的路径。还应该指出的是,Spectralis-score 以百万分之 20 的容差集成了 Prosit 预测,利用了更高分辨率的 m/z 比信息。
该团队还展示了一种罕见错义变异的证据,其最大等位基因频率小于 1%。因此,独立于基因组数据识别罕见变异的能力为光谱包含个人身份信息的想法提供了证据。随着从头肽测序的不断改进,研究人员越来越接近能够通过质谱法重新识别个体。因此,研究人员认为原始质谱蛋白质组数据必须通过数据访问门户共享,并采用与 NGS 测序数据类似的数据控制措施。
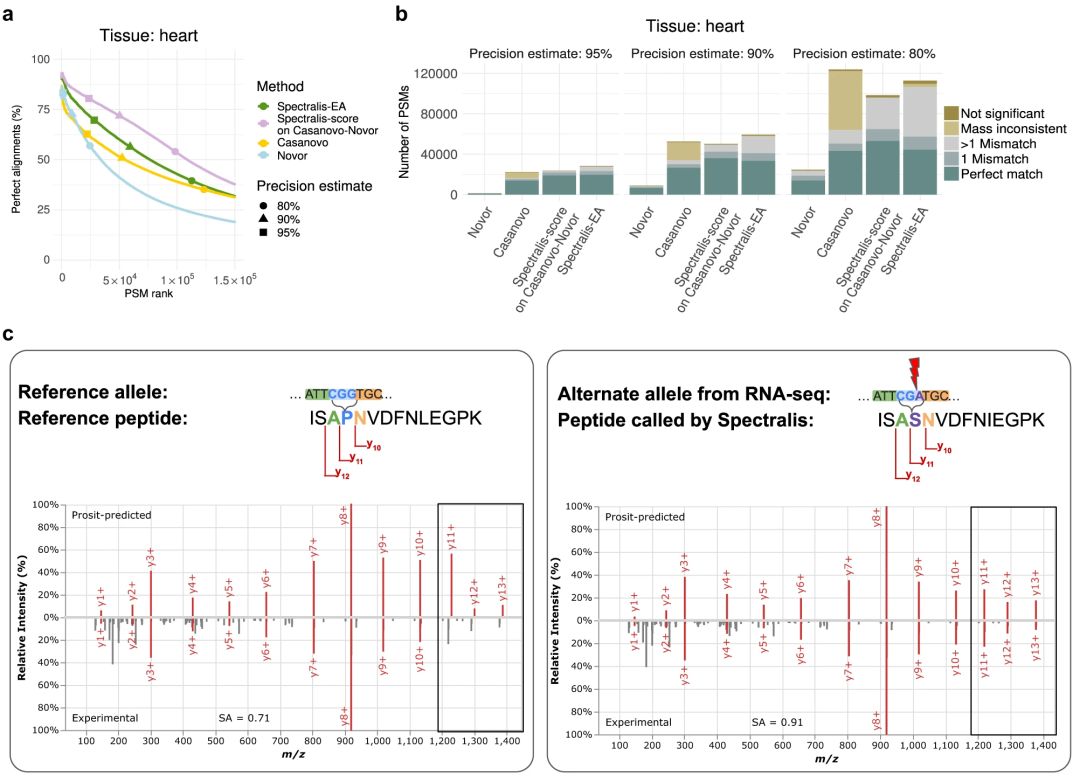
图:应用于未鉴定的光谱和变体调用。(来源:论文)
竞品 Casanovo 的新版本 Casanovo v3.2.0 是与 Spectralis 同时开发的。Casanovo v3.2.0 是 Casanovo v2.0.0 的大幅改进版本,是通过在由约 3000 万 PSM 组成的非常大的数据集上进行训练而获得的。
在 Casanovo v3.2.0 上训练的 Spectralis-score 的修订版得分仍然较低,但在九个物种中的六个物种上以 90% 的精确度显著提高了召回率。
未来的工作有必要进一步研究两种方法的互补性,例如,通过针对 Casanovo v3.2.0 的剩余错误训练 bin 重分类算法。
该研究的一个局限性是 Spectralis 迄今为止仅限于单一翻译后修饰,即蛋氨酸氧化。进一步的翻译后修改可以在未来的工作中通过扩展 AA 间隙卷积来解决。例如,模拟动物磷酸化只需要添加三种氨基酸的磷酸化质量。
另一个限制是该方法假设每个光谱有一个正确的肽。为此,研究人员将数据库搜索基本事实限制为每个光谱最多一个肽。然而,研究估计所有光谱中大约一半是嵌合的,即它们包含来自两个或多个具有相似质量和保留时间的前体离子的峰。
这进一步解释了 Spectralis 和其他早期从头肽测序工具的整体召回率有限,这些工具都假设每个谱图只有一个肽。对肽混合物进行建模需要不同的建模方案并建立合适的地面实况数据。
尽管存在这些限制,Spectralis 仍表现出强大的从头肽测序性能,尤其是在高精度范围内,使其可用于变异识别。因此,它可以使蛋白质组学更适合从病原体监测到免疫肽组学和宏蛋白质组学等应用。
论文链接:https://www.nature.com/articles/s41467-023-44323-7
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










