将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


编辑 | ScienceAI
近日,浙江大学杭州国际科创中心生物与分子智造研究院邢华斌教授团队和陈华钧教授团队瞄准多孔吸附剂材料的精准智造,开发出专家知识共学习的晶态多孔材料吸附性能端对端深度学习框架 DeepSorption,有效提升多孔材料吸附性能的预测精度与速度,并实现了原子尺度的可解释性。
这一成果以《Direct prediction of gas adsorption via spatial atom interaction learning》为题,于 2023 年 11 月 3 日发表在《Nature Communications》上。

论文链接:https://www.nature.com/articles/s41467-023-42863-6基于多孔材料的物理吸附技术为二氧化碳捕获、能源气体储存、化工分离过程等全球挑战提供了低成本和节能环保的解决方案。然而,基于 trial-and-error 实验范式的传统高性能多孔材料的筛选和设计方式受到实验时间长、试错成本高等问题的阻碍。
机器学习通过学习多孔材料及其物理吸附行为的知识,为快速发现具有所需吸附特性的材料提供了一种有效的方法。传统的基于专家设计的多孔材料描述符的机器学习算法通过最大限度地包含多孔材料的关键结构信息来提高模型预测的准确性。然而,由于该方法存在的原始结构信息丢失和描述符生成和处理过程中计算成本高的固有缺点不可避免地会导致误差的传播,造成模型预测精准度的局限。
端到端的深度学习模型有利于保持完整的原始结构信息,具有实现准确预测的巨大潜力。然而,要直接从多孔材料结构出发实现有效的吸附性能预测,还面临三个艰巨的挑战:(1)完整的原始结构信息的输入和传递;(2)原子尺度的信息交互和计算以及良好可解释性;
(3)在纯数据驱动的深度学习模型中存在的数据饥渴问题。
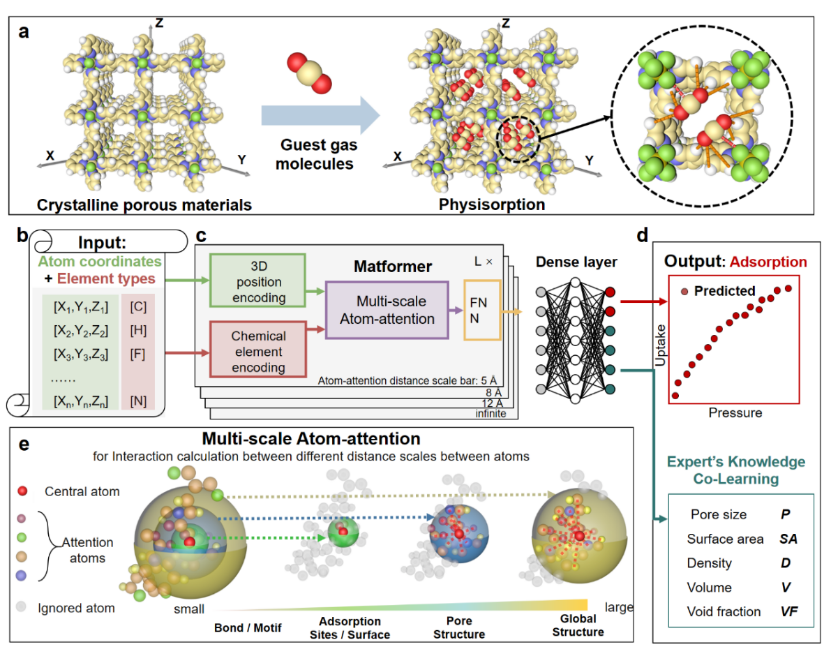
图 1:DeepSorption 深度学习框架的组成部分及多尺度原子注意力机制介绍。本研究设计并训练了一个内置专家知识共学习模块的空间原子相互作用学习框架 DeepSorption,实现了利用晶态多孔材料的原子坐标和化学元素类型的信息作为输入,结构吸附性能的端对端预测。
该框架的独特架构在于开发的 Matformer 模型,该模型可以高保真地解释多孔材料的整体结构信息,包括原子空间排列和化学元素信息。此外,模型内的多尺度原子注意(MSA)机制实现了对不同尺度原子间相互作用的准确、高效认知,并实现了隐藏在编码层中的潜在原子相互作用的可视化。此外为针对纯数据驱动学习中存在的数据饥渴的弊端,采用了专家知识共同学习(knowledge co-learning, KCL)策略。
结果表明,KCL 可以将专家知识作为预测的辅助任务,促进模型在结构-吸附性能构效关系空间中的收敛,有利于提高吸附性质的预测精度。值得注意的是,专家知识的输出只在模型训练时需要,而不干扰预测过程,保证了快速的预测速度。
DeepSorption 深度学习框架拥有时间效率高、误差传播少以及数据效率高的优点。与巨正则蒙特卡罗分子模拟和其他机器学习模型相比,使用DeepSorption 进行晶态多孔吸附剂的气体吸附曲线预测的精度更高、速度更快。与基于专家信息描述符的机器学习模型和晶体图神经网络 CGCNN 相比,DeepSorption 在多个二氧化碳和氮气的吸附量数据集上预测性能的平均绝对误差下降了 20-35%。
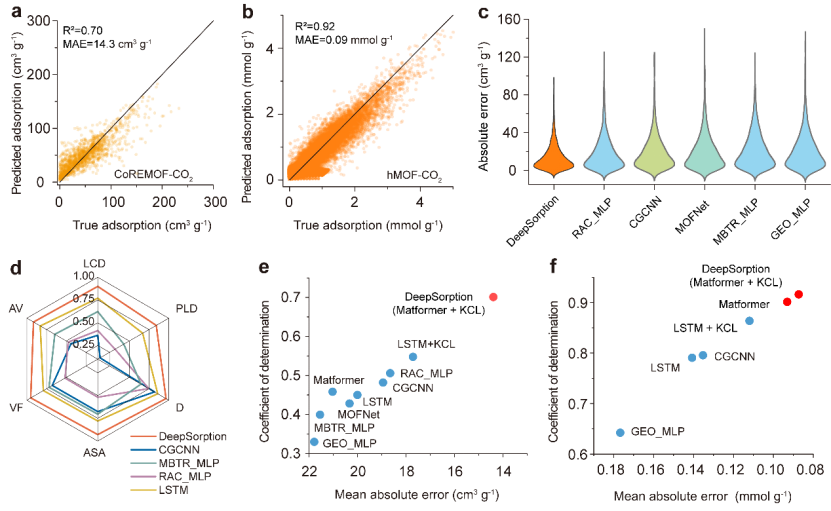
图 2:DeepSorption 在 CoREMOF 和 hMOF 数据集上预测性能和对比。DeepSorption 通过空间原子相互作用学习网络实现了晶体多孔材料复杂吸附特性的准确、快速预测。得益于多尺度原子注意机制,DeepSorption 能够准确评估原子之间的相互作用,实现物理吸附行为预测,并提供直观的可视化思维和执行轨迹。复杂物理化学性质的准确预测凸显了整体结构意识、原子级空间结构信息与化学元素信息的耦合传递和相互作用的重要性。
DeepSorption 深度学习框架不仅实现晶态多孔材料吸附性能的端对端准确预测,并在原子尺度揭示了决定晶体多孔材料表达功能的内在化学性质,并有望成为预测其他晶体材料(如钙钛矿和晶态催化剂)的各种物理化学和表面性质的一种基准算法工具。
文章的第一作者是浙江大学崔稷宇、清华大学吴方、浙江大学软件学院张文研究员和浙江大学化学工程与生物工程学院杨立峰研究员,浙江大学化学工程与生物工程学院邢华斌教授和浙江大学计算机科学与技术学院陈华钧教授为共同通讯作者。
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅
读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










