将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


编辑 | 萝卜皮
细胞类型反卷积是一种用于从大量测序数据中确定/解析细胞类型比例的计算方法,并且经常用于分析肿瘤组织样本中的不同细胞类型。然而,由于重复性/再现性、参考标准可变以及缺乏单细胞蛋白质组参考数据的挑战,使用蛋白质组数据分析细胞类型的反卷积技术仍处于起步阶段。
哈尔滨工业大学、腾讯 AI lab 以及苏黎世联邦理工学院的研究团队合作开发了一种专门针对蛋白质组数据设计的基于深度学习的反卷积方法(scpDeconv)。
scpDeconv 使用自动编码器利用来自批量蛋白质组数据的信息来提高单细胞蛋白质组数据的质量,并采用域对抗架构来桥接单细胞和批量数据分布,并将标签从单细胞数据转移到批量数据 。
该研究以「Deep domain adversarial neural network for the deconvolution of cell type mixtures in tissue proteome profiling」为题,于 2023 年 10 月 19 日发布在《Nature Machine Intelligence》。

亟需处理单细胞蛋白质组数据的新方法
批量测序技术一般是指对特定组织、器官或器官系统中的所有细胞进行裂解和测序,并随后测量基因/蛋白质的平均丰度。因此,批量测序忽略了样品的细胞异质性。单细胞(转录组/蛋白质组)测序技术的发展允许从组织中分离单细胞,以分析和测量各个分子。
尽管该技术在概念上具有优势,但由于成本相对较高且与福尔马林固定或石蜡包埋组织的样本不兼容,应用它来探索大样本队列中肿瘤微环境的细胞异质性一直具有挑战性。因此,人们开发了反卷积方法来从大量组织样本的表达谱中推断细胞类型及其比例,为临床背景下研究肿瘤微环境的细胞组成提供了一种相对低成本和便捷的方法。
然而,到目前为止,现有的反卷积方法仅关注转录组学。尽管蛋白质组提供了从理解生物机制到发现药物靶点的关键生物学和临床信息,但尚未关注对大量蛋白质组数据进行解卷积。
此外,由于复杂的转录后调控、RNA/蛋白质降解和翻译后修饰,大多数蛋白质的浓度无法通过相应基因的转录/表达水平准确表示。大多数蛋白质的拷贝数比相应转录本高 1,000 倍以上,并且表达比转录本更广泛的动态丰度范围,因此在区分细胞和细胞状态并提供独特的生化见解方面表现出更高的潜力。
所以,迫切需要开发基于单细胞蛋白质组数据的新方法来对大量蛋白质组样本进行解卷积。
已有反卷积方法有诸多挑战
迄今为止,最常用的单细胞蛋白质组技术主要基于分子标记抗体,但是检测到的蛋白质数量有限。随着基于质谱的单细胞蛋白质组学技术的出现,单个细胞中检测到的蛋白质数量已大幅增加至多达 1,000-3,000 个细胞内蛋白质,从而扩大了检测到的蛋白质的数量和类型。然而,处理足够数量的细胞来生成数据以支持来自批量数据的细胞类型反卷积仍然具有挑战性。
与转录组数据相比,训练和基准反卷积算法所需的单细胞蛋白质组数据还存在许多额外的挑战。这些包括单细胞蛋白质组数据中的大量背景噪音、较差的数据质量和取决于分析运行和/或技术的显著变化,以及有限的蛋白质组覆盖范围。
此外,现有的针对转录组数据设计的反卷积方法并不直接适用于蛋白质组数据。
首先,蛋白质丰度和转录本表达有不同的分布和值范围。其次,与批量蛋白质组学相比,单细胞蛋白质组学检测到的蛋白质组覆盖率要低得多,这是现有反卷积方法未考虑的情况。第三,对于单细胞蛋白质组数据和批量蛋白质组数据,「批次效应」(分布之间的变异性)很明显,这是现有的反卷积方法很少考虑的。
基于深度学习的方法 scpDeconv
为了应对这些挑战,通过充分考虑最近开发的单细胞蛋白质组技术的固有特征,哈尔滨工业大学、腾讯 AI lab 以及苏黎世联邦理工学院的研究团队开发了一种专门针对蛋白质组数据定制的新的反卷积方法——scpDeconv。
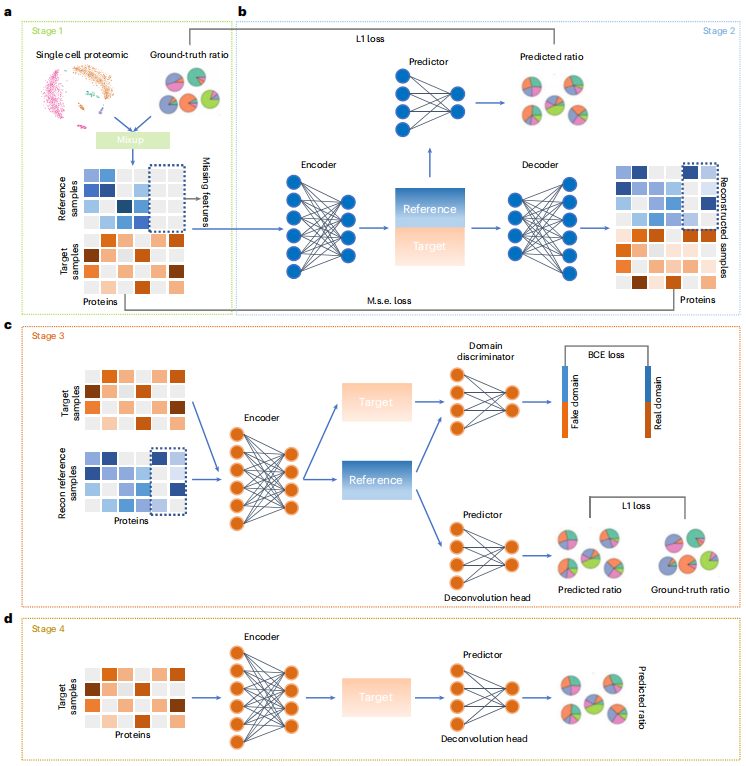
图示:scpDeconv 方法的架构。(来源:论文)通过使用自动编码器网络,scpDeconv 的插补模块可以插补单细胞蛋白质组数据中缺失的低丰度蛋白质的值。这是通过对混合的单细胞蛋白质组数据和目标批量蛋白质组数据的联合学习来实现的。通过领域对抗训练,scpDeconv 可以提取估算参考数据的领域不变潜在特征并将标签转移到目标数据。
该团队严格而广泛的实验评估了 scpDeconv 在细胞类型反卷积以及从大量组织蛋白质组数据集中推断细胞周期状态方面的性能和鲁棒性。此外,对临床黑色素瘤样本中现有的大量蛋白质组数据集的重新分析显示了 scpDeconv 在癌症诊断和预后中的临床应用价值。
scpDeconv 的优势
与现有的针对转录组学的反卷积方法相比,scpDeconv 为蛋白质组数据反卷积提供了四个级别的优势。
(1)scpDeconv 以数据驱动的方式基于参考数据和目标数据的联合学习来推断细胞类型比例,因此不受先前数据分布假设的限制,并且与来自不同实验或技术背景的广泛蛋白质组数据兼容。
(2)scpDeconv引入了域适应策略,可以有效地处理批次效应并缩小参考数据和目标数据之间的分布差距。
(3)scpDeconv 可以在插补模块的帮助下插补单细胞蛋白质组数据中低丰度蛋白质的缺失值,并提高参考数据的质量。
(4)研究人员验证了scpDeconv的输出能够反映肿瘤微环境中的细胞类型分布,并且由此确定的细胞类型分布对于疾病诊断和预后有意义。
局限性与展望
然而,scpDeconv 也显示出一些局限性。模型训练所需的具有已知细胞类型组成的参考蛋白质组数据,是由具有标记细胞类型的单细胞蛋白质组数据构建的。目前,单细胞蛋白质组学仍处于起步阶段,尚未应用于广泛的生物样品。该研究中详尽收集的单细胞蛋白质组数据仅涉及少数细胞类型,从而限制了 scpDeconv 目前的适用性。
该算法专为使用单细胞蛋白质组数据作为参考从蛋白质组数据中进行细胞类型/状态解卷积而定制,将具有许多关键应用。这些包括细胞类型的反卷积、细胞状态的估计(以细胞周期状态为例)以及对多种癌症的微环境的重新分析,从而为已经获得的大量蛋白质组数据集添加额外的值。
因此,研究人员预计 scpDeconv 将支持从大量蛋白质组数据中提取额外的、生物学和临床上重要的结果,从而使蛋白质组学界受益。随着对公开的单细胞蛋白质组图谱的探索,未来在更广泛的背景下重新分析现有的组织蛋白质组数据将是可行的。此外,凭借一系列针对多种癌症类型的可靠微环境参考,相信 scpDeconv 将能够以低成本分析某些肿瘤样本的所有成分,从而有利于基础医学研究。
论文链接:https://www.nature.com/articles/s42256-023-00737-y
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










