将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


编辑 | 萝卜皮
合理设计具有所需生物活性的新型分子是药物发现中一项关键但具有挑战性的任务,特别是在治疗新靶标家族或未充分研究的靶标时。
中南大学的研究团队提出了一种药效团引导的深度学习方法来生成生物活性分子(PGMG)。通过药效基团的指导,PGMG 为生成生物活性分子提供了灵活的策略。
PGMG 使用图神经网络对空间分布的化学特征进行编码,并使用 Transformer 解码器来生成分子。引入潜在变量来解决药效团和分子之间的多对多映射,从而提高生成分子的多样性。
与现有方法相比,PGMG 生成的分子具有很强的对接亲和力,并且有效性、独特性和新颖性得分很高。
该研究以「A pharmacophore-guided deep learning approach for bioactive molecular generation」为题,于 2023 年 10 月 6 日发布在《Nature Communications》。

获取生物活性化合物是药物发现中至关重要但具有挑战性的一步。据估计,对于遵守 Lipinski 的「五规则」的分子,类药物化学空间高达 10^60,这是一组根据其分子特性评估化合物作为口服活性药物的潜力的标准。因此,在如此巨大的空间中寻找所需的分子是极其困难的。
传统上,对特定靶标表现出初始活性的命中化合物可以从药物化学家设计的天然产物中获得或通过高通量筛选获得。这些方法消耗大量的人力和财力,使得热门化合物的获取效率低下且成本高昂。
最近,人们提出了深度生成模型来合理设计具有所需特性的新型分子,为这项任务提供了新的视角。在从深度神经网络生成分子的流行架构和模型中,变分自动编码器、强化学习、生成对抗网络和自回归模型已经成功地在指定的前提下设计了所需的分子。许多方法旨在生成具有给定物理化学性质的分子,例如 Wildman-Crippen 分配系数(LogP)、合成可及性(SA)、分子量(MW)和药物相似性定量估计(QED)。
然而,更实际和更具挑战性的目标是设计满足涉及生物实验或大量计算来近似的特性的分子,例如分子针对特定靶标的生物活性。为了生成生物活性分子,现有模型需要大量已知活性分子的数据集进行微调。但是,该数据集可能不可用。
活性数据的缺乏是在药物设计中应用基于深度学习的方法的主要障碍之一,特别是对于新的靶标家族。药物设计策略的选择还取决于可以使用哪些信息,例如受体结构或一些已知的活性配体,从而缩小了许多深度学习方法的应用范围。
在最新的研究中,中南大学的研究团队提出了药效团引导的生物活性分子生成深度学习方法(PGMG),这是一种基于深度学习的药效团引导的分子生成方法。
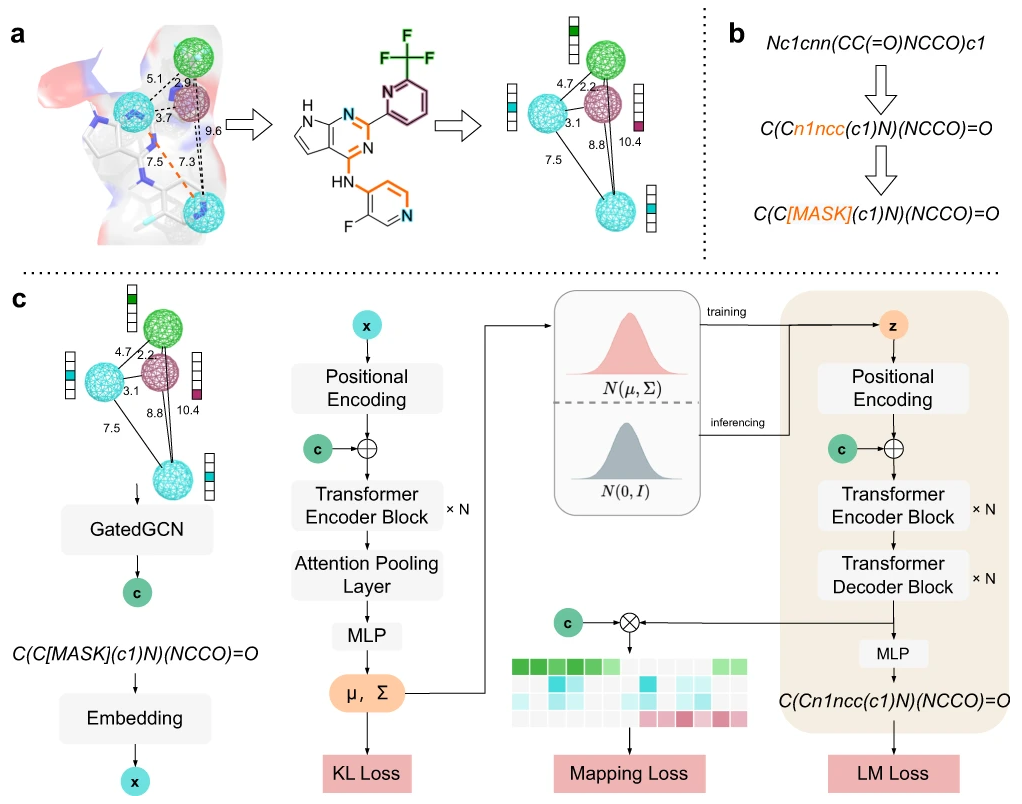
图示:PGMG的整体架构。(来源:论文)
PGMG 使用药效团假设作为连接不同类型活动数据的桥梁。药效团是药物与靶标结合所必需的一组空间分布的化学特征。药效团假设可以通过叠加一些活性化合物来构建,也可以从给定靶标的结构中推断出来。基于药效团的药物设计有许多成功的应用,但其在深度生成模型中的潜力尚未得到充分开发。
PGMG 可以在不同的药物设计场景中实现灵活的生成,而无需进一步微调,特别是对于活性数据不足的新发现的靶点。研究人员用完整的图来完整地表示一个药效团,每个节点对应一个药效团特征,这样空间信息就可以编码为每个节点对之间的距离。使用图表作为唯一输入,PGMG 可以生成与相应药效团匹配的分子。这使得 PGMG 能够以统一的表示形式和具有生物学意义的方式利用不同类型的活动数据来控制分子设计的过程。
此外,由于药效团和分子具有多对多的关系,PGMG 引入了潜在变量来建模这种关系,以增加生成分子的多样性。并且,他们采用 transformer 结构作为骨干来学习 SMILES 字符串的隐式规则,从而在潜在变量和分子之间进行映射。研究人员通过目标导向和类药物指标全面评估 PGMG 在分子生成方面的性能。结果表明 PGMG 能够生成大量分子,其对接分数与从 ChEMBL 数据库获得的已知活性分子相似甚至更好。通过针对某些靶点的药效团,PGMG 还可用于设计双靶点或多靶点分子。
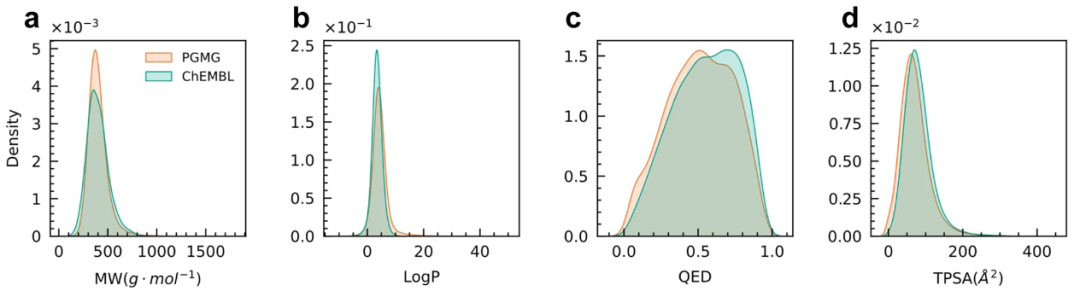
图示:ChEMBL 训练集和 PGMG 生成的分子的理化特性分布。(来源:论文)
另外,研究人员期望可以采用 PGMG 来制备化学库以提高虚拟筛选效率,因为这可以为特定靶点提供一定数量的候选药物样分子。基于结构和配体的案例研究表明,PGMG 可以生成与药效团假说相匹配且具有结构多样性的高质量生物活性分子,这表明 PGMG 可以应用于多种药物设计场景,例如研究替代医学和耐药性。最后,支架跳跃的展示表明 PGMG 可以发现具有新颖支架的活性分子。
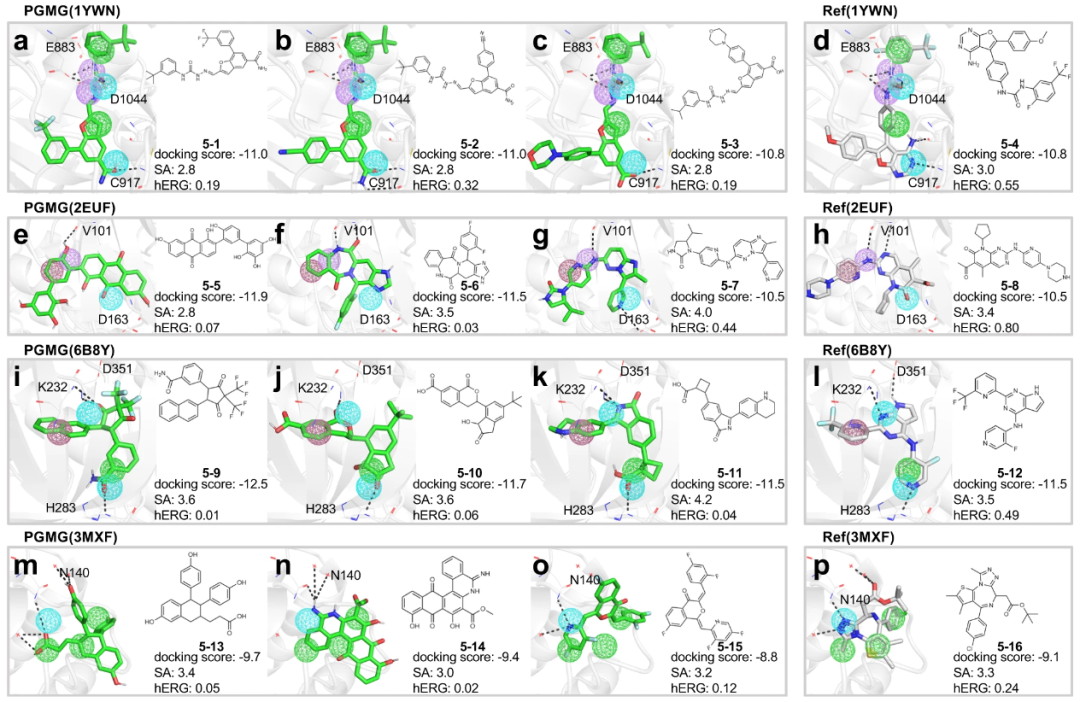
图示:基于结构的药物设计中 PGMG 生成的分子的结合位点。(来源:论文)从头药物设计是一个复杂且针对具体情况的问题,计算方法应受益于先前生物化学知识的输入。PGMG 通过将药效团假设的构建留给用户来受益于这一想法。形成药效团假说的方法有多种;可以使用各种信息,并且可以进行不同的调整来增强假设。可以使用更准确的假设来导入生成的分子的质量。例如,QSAR 研究可用于调整假设,这可能会对生成的分子产生更好的约束。另一方面,应该承认 PGMG 的局限性。例如,PGMG 目前不支持药效团假设中的排除体积。由于这里研究人员专注于生成具有所需活性的分子的任务,因此 PGMG 并没有明确限制生成的分子的属性。
该团队未来的工作方向是在 PGMG 中包含排除体积和其他功能,使生成的分子更加可控和可塑。另一个方向是将 QSAR 分析纳入生成模型,这可以为药效团假设提供更强的基线,并增强可解释性。
PGMG服务器:https://www.csuligroup.com/PGMG
论文链接:https://www.nature.com/articles/s41467-023-41454-9
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










