将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

编辑 | 绿萝
物质中电子的排列(称为电子结构)在药物设计和能量存储等基础研究和应用研究中发挥着至关重要的作用。不同应用的建模和模拟主要依赖于密度泛函理论(DFT),它已成为预测物质电子结构的主要方法。
虽然 DFT 计算非常有用,但其计算尺度限制了它们只能用于小型系统。
近日,来自德国 Helmholtz-Zentrum Dresden-Rossendorf(HZDR)研究所的高级系统理解中心 (CASUS) 和美国桑迪亚国家实验室的研究人员开发了一种基于机器学习的电子结构预测模拟方法——材料学习算法(Materials Learning Algorithms,MALA),用于预测任何长度尺度上的电子结构。
MALA 通过将机器学习与物理算法相结合,为小型系统提供超过 1,000 倍的加速。其可以在 DFT 计算不可行的规模上进行预测,能够精确模拟超过 100,000 个原子的大型系统,优于传统方法。这项创新将彻底改变应用研究,并且与高性能计算系统高度兼容。
论文的通讯作者、CASUS 的 Attila Cangi 表示:「我们认为 MALA 将引发电子结构计算的变革,因为我们现在有一种方法可以以前所未有的速度模拟更大的系统。」
该研究以「Predicting electronic structures at any length scale with machine learning」为题,于 2023 年 6 月 27 日发布在《npj Computational Materials》上。
论文链接:https://www.nature.com/articles/s41524-023-01070-z电子作为重要的基本粒子。它们彼此之间以及与原子核之间的量子力学相互作用,产生了化学和材料科学中观察到的多种现象。了解和控制物质的电子结构可以深入了解分子的反应性、行星内的结构和能量传输以及材料失效的机制。
计算建模和模拟、高性能计算越来越多地用来解决科学挑战。然而,由于缺乏将高精度与跨不同长度和时间尺度的可扩展性结合起来的预测建模技术,实现量子精度的真实模拟存在障碍。
经典原子模拟方法可以处理大型复杂系统,但其对量子电子结构的忽略限制了其适用性。相反,不依赖于经验建模和参数拟合(第一原理方法)等假设的模拟方法提供了高保真度,但计算要求较高。例如,DFT 是一种广泛使用的第一原理方法,DFT 研究对象通常包含多个电子,而粒子数大于等于三个的力学模型是无法精确求解的。从而将其预测能力限制在小尺度上。
基于深度学习的混合方法
研究人员团队现在提出了一种新颖的模拟方法,称为 MALA 软件堆栈。在计算机科学中,软件堆栈是算法和软件组件的集合,它们组合在一起创建用于解决特定问题的软件应用程序。
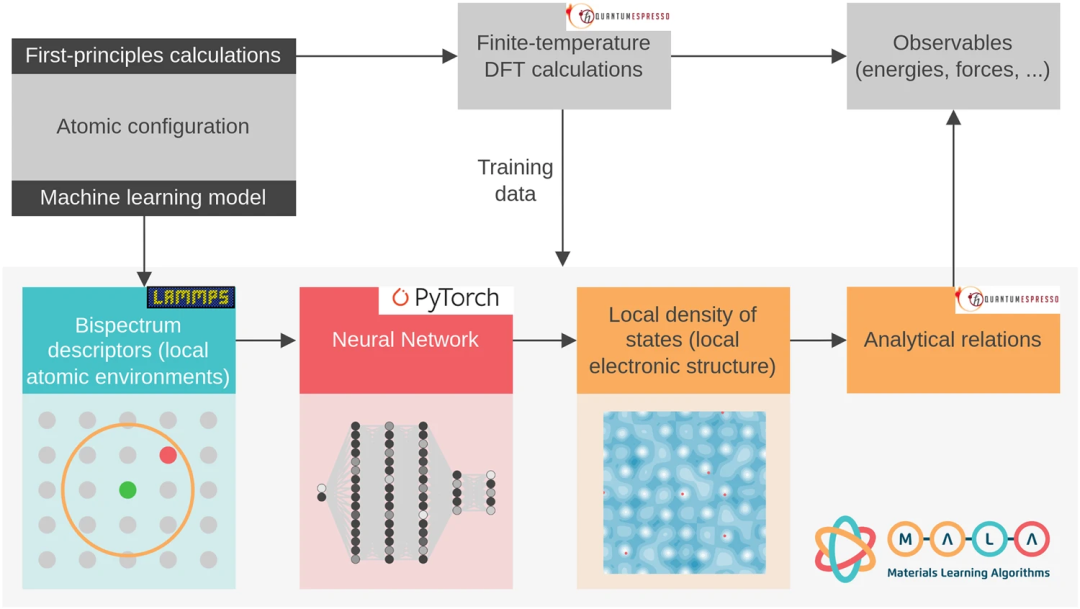
MALA 的各个步骤如图 1 所示。它们包括结合双谱描述符的计算来编码原子密度、训练和评估神经网络来预测 LDOS,最后将 LDOS 后处理为物理可观测值。整个工作流程作为 MALA 软件包端到端实现,研究使用流行的开源软件包的接口,即 LAMMPS(描述符计算)、PyTorch(神经网络训练和推理) 和 Quantum ESPRESSO(将电子结构数据后处理为可观测值)。
MALA 的主要开发人员、CASUS 的博士生 Lenz Fiedler 解释说:「MALA 将机器学习与基于物理的方法相结合来预测材料的电子结构。它采用了一种混合方法,利用一种称为深度学习的既定机器学习方法来准确预测局部量,并辅以物理算法来计算感兴趣的全局量。」
MALA 软件堆栈将空间中原子的排列作为输入,并生成称为双谱分量的指纹,对笛卡尔网格点周围原子的空间排列进行编码。MALA 中的机器学习模型经过训练,可以根据该原子邻域预测电子结构。MALA 的一个显著优势是其机器学习模型能够独立于系统规模,使其能够根据小型系统的数据进行训练,并在任何规模上部署。
精确模拟超 100,000 个原子的大型系统
研究人员通过计算包含超过 100,000 个原子的材料样本的电子结构来证明其工作流程的有效性。
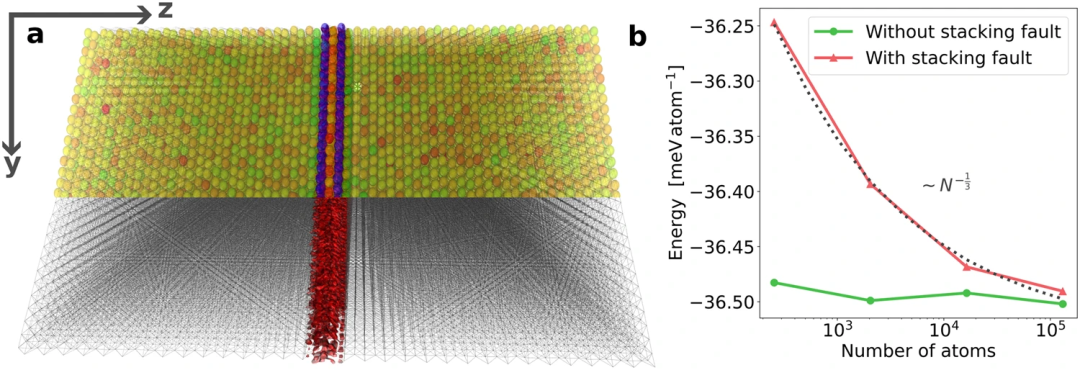
图 2:MALA 模型的尺寸可转移性。(来源:论文)研究人员对 131,072 个原子系统的 ML 预测在 150 个标准 CPU 上仅需要 48 分钟。MALA 能够在超过 100,000 个原子的大规模电子结构中进行精确计算。所提出的 ML 工作流程的计算成本比传统的 DFT 计算低几个数量级,其规模为 ~ N^3。
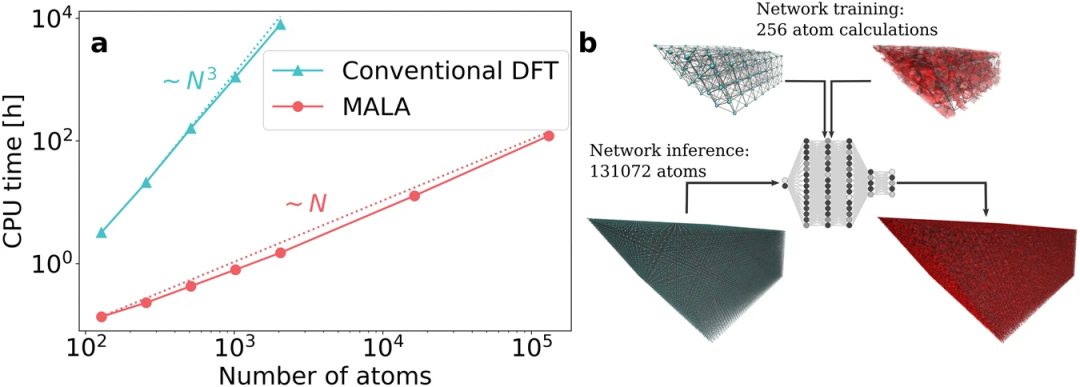
Cangi 解释道:「随着系统规模的增大和涉及的原子数量的增加,DFT 计算变得不切实际,而 MALA 的速度优势不断增强。MALA 的关键突破在于其 「能够在局部原子环境中运行,从而实现精确的数值预测,并且受系统尺寸的影响最小。这一突破性的成就开启了曾经被认为无法实现的计算可能性。」
预计将推动应用研究
未来,研究人员将能够在显著改善的基线基础上解决广泛的社会挑战,包括开发新的疫苗和新型储能材料,对半导体器件进行大规模模拟,研究材料缺陷,探索将大气温室气体二氧化碳转化为气候友好型矿物的化学反应。
此外,MALA 的方法特别适合高性能计算 (HPC)。随着系统规模的增长,MALA 可以在其使用的计算网格上进行独立处理,从而有效地利用 HPC 资源,特别是图形处理单元。
桑迪亚国家实验室的科学家兼并行计算专家 Siva Rajamanickam 解释说:「MALA 的电子结构计算算法很好地映射到具有分布式加速器的现代 HPC 系统。MALA 具有分解工作和在不同加速器上并行执行不同网格点的能力,这使得它成为高性能计算资源上可扩展机器学习的理想匹配,从而在电子结构计算中实现无与伦比的速度和效率。」
参考内容:
https://scitechdaily.com/electronic-renaissance-how-machine-learning-reimagines-material-modeling/
https://phys.org/news/2023-07-machine-enables-accurate-electronic-large.html
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










