理论计算在材料领域的作用已经被越来越多的研究人员所认可,近年来,随着“计算+实验”模式的确定,DFT计算发文量暴涨,逼近每年3万篇。计算模拟不仅成为Science、Nature等顶刊标配,更逐步变为顶刊必备!
但DFT计算具有一定的技术壁垒,大多数科研人员会面临同样的困惑:
DFT计算有什么用?该用什么软件?如何搭建模型,怎样分析结果?我算的到底对不对?审稿人质疑我怎么办?
为了解决大家的疑惑,华算科技推出DFT计算特训营,朱老师带大家一步步入门DFT计算,早发顶刊,顺利毕业!课程形式:12小时直播,价值1280元,限时免费、提供回放。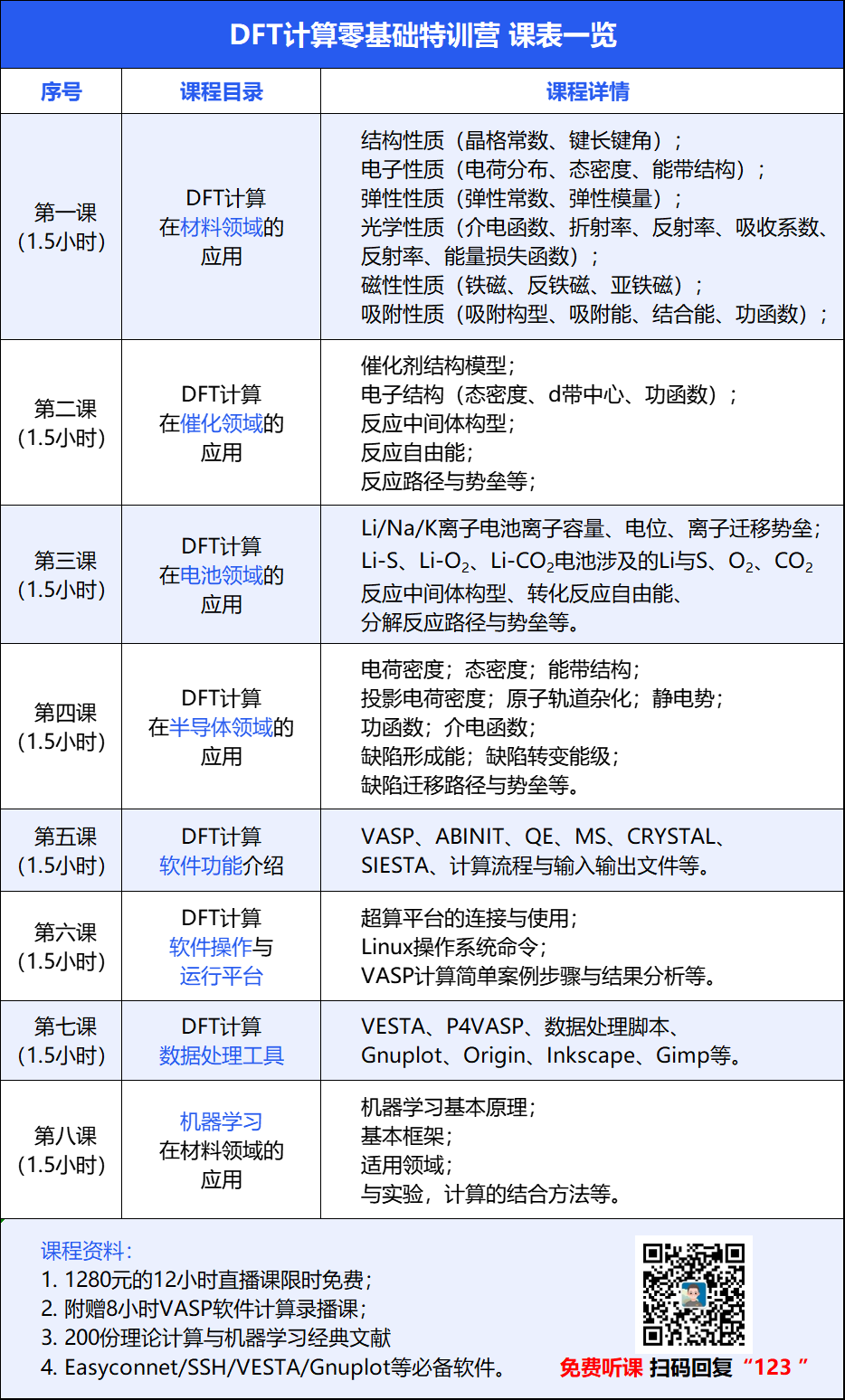
扫描下方二维码,回复“123”免费听课、下载资料。朱老师,同济大学本科直接攻读博士学位(4年),海外3年以上博后经历,发表高质量独立一作SCI论文30篇。
回国后被授予深圳市海外高层次人才,拥有14年VASP重度使用经验,成功讲授100+场VASP计算培训和超过6W人的学习理论计算公开课。第一期
主要介绍DFT计算所能得到的结构(晶格常数、键长键角)、电子(电荷分布、态密度、能带结构)、弹性(弹性常数、弹性模量)、光学(介电函数、折射率、反射率、吸收系数、反射率、能量损失函数)、磁性(铁磁、反铁磁、亚铁磁)、吸附(吸附构型、吸附能、结合能、功函数)等性质与实验结果结合的文献案例,说明DFT计算在各种材料研究领域的应用。晶体表面吸附分子与自旋密度
第二期
主要介绍DFT计算所能得到的催化剂结构模型、电子结构(态密度、d带中心、功函数)、反应中间体构型、反应自由能、反应路径与势垒等计算结果与实验结果结合的文献案例,说明如何在电催化领域运用DFT计算。CO2RR自由能与中间体构型
主要介绍DFT计算所能得到的Li/Na/K离子电池离子容量、电位、离子迁移势垒,Li-S、Li-O2、Li-CO2电池涉及的Li与S、O2、CO2反应中间体构型、转化反应自由能、分解反应路径与势垒,说明如何在电池领域运用DFT计算。
多硫化锂吸附与转化自由能
第四期
主要介绍DFT计算所能得到的电荷密度、态密度、能带结构、投影电荷密度、原子轨道杂化、静电势、功函数、介电函数、缺陷形成能、缺陷转变能级、缺陷迁移路径与势垒等计算结果与实验结果结合的文献案例,说明如何在半导体领域运用DFT计算。半导体缺陷形成能
第五期
主要介绍VASP、ABINIT、QE、MS、CRYSTAL、SIESTA等常见DFT计算软件的优势与不足,并简要介绍VASP计算流程与输入输出文件,说明在不同研究领域如何选择正确DFT计算软件以达到实验与理论结合的目的。VASP wiki
第六期
主要介绍超算平台的连接与使用、Linux操作系统命令、VASP计算简单案例步骤与结果分析,让实验人员了解DFT计算的具体的工作内容以及如何分析计算结果。Li在二维材料表面吸附
第七期
主要介绍VASP计算结果的处理工具,包括VESTA、P4VASP、数据处理脚本、Gnuplot、Origin、Inkscape、Gimp,让实验人员了解如何处理DFT计算数据以实现文献中的图片效果。
P4VASP软件
第八期
主要介绍机器学习在材料领域的使用方法,包括机器学习基本原理,基本框架,适用领域,与实验,计算的结合方法,让化学与材料学的研究人员能读懂机器学习相关文献,并了解如何在自己的领域中使用机器学习。








