
近年来,理论计算与机器学习,越来越受顶刊青睐!“实验+计算”的模式已成为顶刊标配,机器学习亦逐渐成为一种强有力的研究工具!
经过1000余天的打磨和改进,华算科技陆续推出VASP计算、Materials Studio计算、机器学习与材料等培训,备受好评,往期学员已陆续在Nature子刊、Angew.、JACS、AM等顶级期刊发表文章!
线上课程,不限时间、不限地点、随报随学、无限次回看。
课程群永不解散,随时提问,及时解答。提供超算练习账号,边学边练。
报名方式:添加下方微信好友,或者联系手机131-2872-3011 。带你零基础入门DFT计算/机器学习,提升论文档次,发顶刊快人一步!学员已发表Nature、Nature Energy、Nature Materials、Angew..、JACS、AM、AEM等期刊。课程内容涉及晶体与二维材料材料的建模、弹性、电子(电荷态、态密度、轨道杂化、能带、功函数)、光学、磁性、吸附、催化性质计算,帮助大家快速理解模型构建与VASP输入文件中各个参数的意义,提高学习效率,完成DFT计算入门。通过VASP软件的晶体结构、电子、弹性、磁学性质,二维材料的结构、吸附、扩散性质的计算练习,帮助大家快速理解模型构建与VASP输入文件中各个参数的意义,提高学习效率,完成DFT计算入门。课程内容涉及晶体表面/二维结构/一维结构的结构性质,功函数、d带中心、吸附能、差分电荷密度等电子性质,HER、OER/ORR、NRR、CO2RR等催化反应的自由能与过渡态计算,CO、CO2、CH4、C6H6等分子的吸附与分解过程。
单原子、双原子催化剂是目前电催化领域研究的最多的催化剂材料,他们具有原子利用率高、活性高、性能精确可调的特点,能够有效地催化各种化学反应,包括HER、OER、CO2RR、NO3RR等,在能源、化工等领域起着重要作用。DFT计算能够深入研究单原子催化剂结构稳定性、电子结构、对反应中间体的吸附能力、以及对反应热力学和动力学势垒的影响,已经被广泛应用于催化剂设计、筛选、性能分析等方面。本次课程由华算科技朱老师主讲单原子催化计算,课程具体涉及催化剂模型,电子性质,HER、OER/ORR、CO2RR、NO3RR等反应的自由能与过渡态计算。
课程具体涉及:理论知识、二维/异质结构建模、能带、功函数、静电势、吸附位点、吸附能、结合能、虚频消除、态密度、d带中心、过渡态、CO2RR、OER/ORR、电位、容量、迁移势垒。课程涉及电池中的电化学反应类型(插入、合金、转化)的判断,正负极材料(三维、二维、二维异质结构)的掺杂、离子容量、离子扩散能垒计算,离子与正负极材料的相互作用机理计算,燃料电池所涉及的H2氧化与O2还原过程决速步、过电位的计算。
本次课程将介绍二维材料、一维材料、二维材料异质结构的结构、缺陷、电子、电池、吸附与催化性质。运用VASP软件计算三维和二维半导体材料的能带、态密度、缺陷形成能、扩散势垒、吸附能、电荷转移性质。
内容涉及建构建模、电荷密度、态密度、能带结构、缺陷形成能、缺陷转变能级、缺陷扩散路径与势垒、吸附能、结合能、差分电荷密度计算。
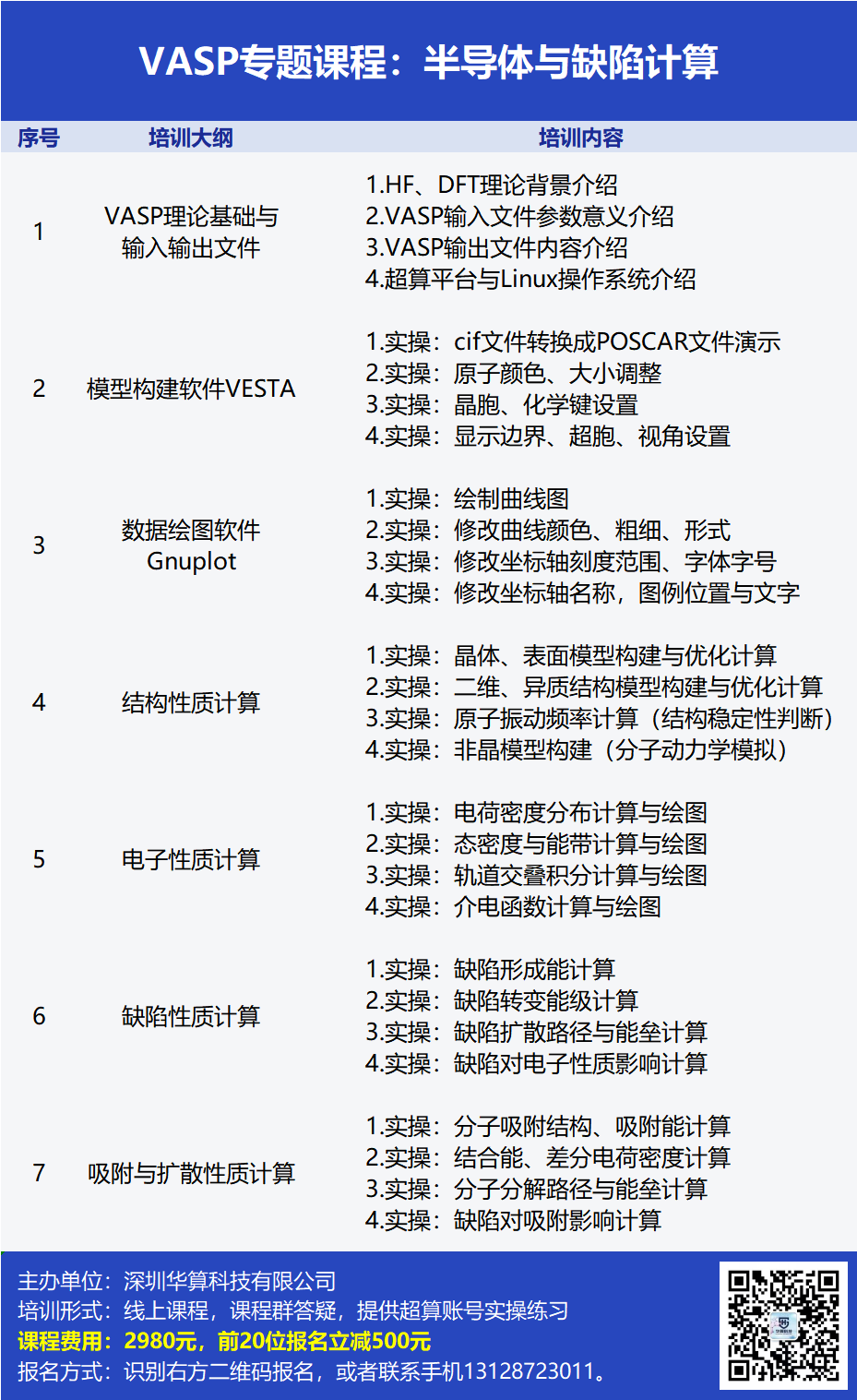
VASP是目前最流行的DFT计算软件,擅长计算半导体材料的结构性质、电子结构、吸附性质。本次课程将运用VASP软件计算钙钛矿半导体的结构、电子、吸附、缺陷性质,二维半导体的结构与电子性质。课程内容将根据学员反馈不断更新。
华算科技Materials Studio零基础培训,0基础起步,32小时高强度训练,27章节理论+实际操作!累计超过1000名学员参与,往期学员已发表Angew.、AM、AEM、Nature子刊等顶刊论文。课程由全职技术专家杨老师主讲,专为初入门学员设计,沿着理论讲解、模型搭建、性质计算、结果分析层层递进。带你学会DFT计算,早发顶刊!华算科技MS杨站长将通过28小时高强度实操培训,基于Materials Studio软件CASTEP、DMol3模块,将HER/OER/ORR/CO2RR台阶图、火山图、d带理论、过渡态、带隙工程、能带电位匹配等催化计算核心问题讲透彻,使大家能将DFT计算用到自己的文章中。科研热点材料:钙钛矿太阳能电池、Janus异质结、二维材料、金属半导体接触、气敏半导体
手段:旋轨耦合SOC、杂化泛函、DFT+U、DFT+D色散矫正、偶极修正
性质分析:载流子迁移率、有效质量、光吸收、能带态密度、能带弯曲、COHP、内建电场、功函数、静电势、整流效应、相变、声子
课程针对编程零基础学员,并包含实验与计算中常见的科研数据处理案例。同学们学习完成后可直接将Python应用于自己的研究数据后处理之中,适合有数据分析需求的实验课题组/计算课题组同学。课程面向Python零基础,对机器学习感兴趣,想在自己的研究方向使用机器学习的化学、材料学相关工作者。通过本次课程,大家可以学会当下最流行的Python语言,学会抓取数据库,能使用机器学习基本算法,并会用于机器学习解决化学与材料学的实验数据处理、材料筛选与性质预测等问题,能够重现机器学习的文献案例。

课程包括:机器学习概念,决策树/贝叶斯/支持向量机/神经网络/随机森林,回归/分类/聚类算法,模型评价/性能度量,小分子/MOF/d带中心/合金催化/单原子催化/钢铁强度/CO2RR/钙钛矿/电极涂层/STM图像,数据库/高通量筛选/Materials Project。本次课程主要基于Python语言与TensorFlow/Keras框架,学习神经网络的构建、训练与使用,课程中使用的绝大部分案例均源于化学/材料文献中真实案例,同学们学习完成后可直接将其用于自己的研究当中。课程设计目的为学习完成之后,能自己构建常用的神经网络,进行各种技巧处理,并进行应用。
主办单位:深圳华算科技有限公司(拥有VASP、Materials Studio、Gaussian、LAMMPS商业版权)
培训形式:课程已上架腾讯课堂,可无限次回看。课程群永不解散,随时提问,及时解答。课程费用:咨询华算科技-小硕,提供增值税普通发票及邀请函。同时报名两个课程可优惠1600元,更多优惠咨询可咨询华算科技-小硕。报名方式:添加下方微信好友报名,或者联系手机131-2872-3011 。朱老师:华算科技资深技术,同济大学本科直接攻读博士学位(4年),海外3年以上博后经历,发表高质量独立一作SCI论文30篇,回国后被授予深圳市海外高层次人才。
拥有14年VASP重度使用经验,成功讲授100+场VASP计算培训和超过10万人的学习理论计算公开课。
杨老师:华算科技全职技术资深专家,深圳市孔雀计划海外高层次人才。曾就职于德国马克思普朗克研究所,日本WPI研究所,并曾在芬兰阿尔托大学进行长期访问,作为PI主持欧盟、日本科研项目6项。拥有10年以上Materials Studio软件使用经验,主要从事固态相变的第一性原理研究、电化学固液界面的AIMD研究与超分子化学中的分子动力学模拟。黄老师:华算科技全职技术专家,武汉大学本科,北京大学博士,新加坡国立大学访问学者。目前已发表SCI文章共20篇,其中第一作者文章5篇,单篇最高影响因子>40。从事理论计算与实验化学研究工作11年,擅长使用机器学习进行化学理论的研究及实验数据的处理,曾获华中地区数学建模邀请赛三等奖,北京大学游戏AI对抗全国邀请赛第四名等相关奖项。











