前面我们演示了 一个完美的单细胞亚群随机森林分离器是如何炼成的,以及 LASSO回归也可以用来做单细胞分类 的两个机器学习算法可以用来做单细胞分类器,而且效果杠杠的。而且也尝试了多种机器学习的算法,比如:不输于LASSO的SVM单细胞分类器
无论是随机森林,LASSO回归,还是支持向量机, 他们的模型都是有点抽象,不容易直观的可视化解释清楚。但是接下来我们要介绍的决策树模型,就不一样。
训练决策树模型
首先,复制粘贴前面的 一个完美的单细胞亚群随机森林分离器是如何炼成的 ,就可以把单细胞表达量矩阵划分为训练集和测试集,然后简单的安装和加载 rpart 包,运行里面的 rpart 函数即可:
library('rpart.plot')
library('rpart')
library('survival')
df = cbind(target=target,as.data.frame(predictor_data))
head(colnames(df))
multivariate 1],collapse = '-')
s 'target ~ ', multivariate )
s # as.formula(s)
fit data=df, method="class" )
summary(fit)
save(fit,file = 'rpart_output.Rdata')
可以看到,我们输入的表达量矩阵是 2000 个基因在接近 2000个细胞里面的表达量信息:
> dim(predictor_data)
[1] 1977 2000
> predictor_data[1:4,1:4]
ISG15 CPSF3L MRPL20 ATAD3C
AAACATACAACCAC -0.8353028 -0.2741145 1.5056616 -0.04970561
AAACATTGAGCTAC -0.8353028 -0.2741145 -0.5625993 -0.04970561
AAACATTGATCAGC 0.3922351 -0.2741145 1.2450489 -0.04970561
AAACCGTGCTTCCG 2.2197621 -0.2741145 -0.5625993 -0.04970561
我们的决策树模型就是把这2000个基因组合一下,来划分细胞的分类。我们简单的可视化一下这个效果:
library(rpart.plot);
rpart.plot(fit, branch=1, branch.type=2, type=2, extra=102,
shadow.col="gray", box.col="green",
border.col="blue", split.col="red",
split.cex=1.2 )
如下所示,虽然是有6个单细胞亚群,但是只需要5个基因就足以区分它们了 :
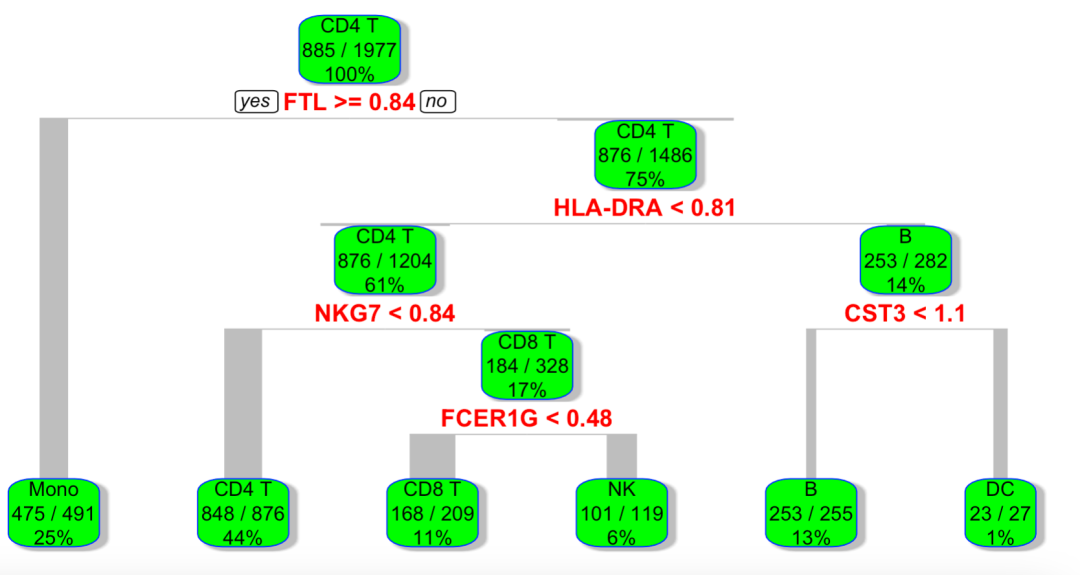
5个基因就足以区分它们了在测试集看模型效果
同样的,训练好的模型,也需要在另外一个数据集看看效果:
test_outputs = predict(fit, as.data.frame(test_expr))
head( test_outputs )
pred_y = colnames(test_outputs)[apply(test_outputs, 1, which.max)]
pred_y = factor(pred_y,levels = levels(test_y))
pdf('rpart-performance.pdf',width = 10)
gplots::balloonplot(table(pred_y,test_y))
dev.off()
可以看到, 容易出问题的仍然是CD8和NK细胞的混入,以及CD4和CD8的混入,这个目前来说是无解的:
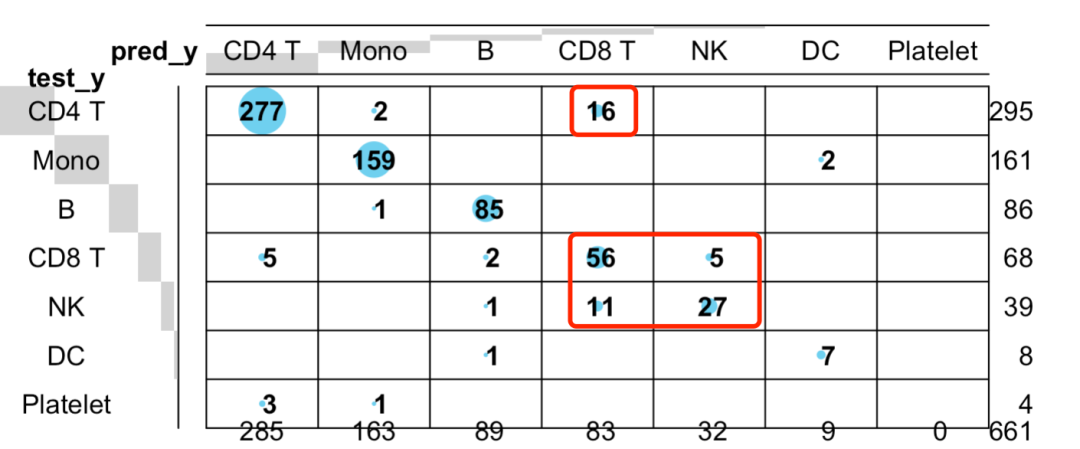
CD8和NK细胞的混入我们可以简单的可视化前面的决策树模型的5个基因:
library(Seurat)
sce=CreateSeuratObject(counts = t(predictor_data) )
Idents(sce) = target
cg = c('FTL','HLA-DRA','NKG7','FCER1G','CST3')
VlnPlot(sce,features = cg ,
stack = T)
ggplot2::ggsave('rpart_models_VlnPlot.pdf' )
如下所示:
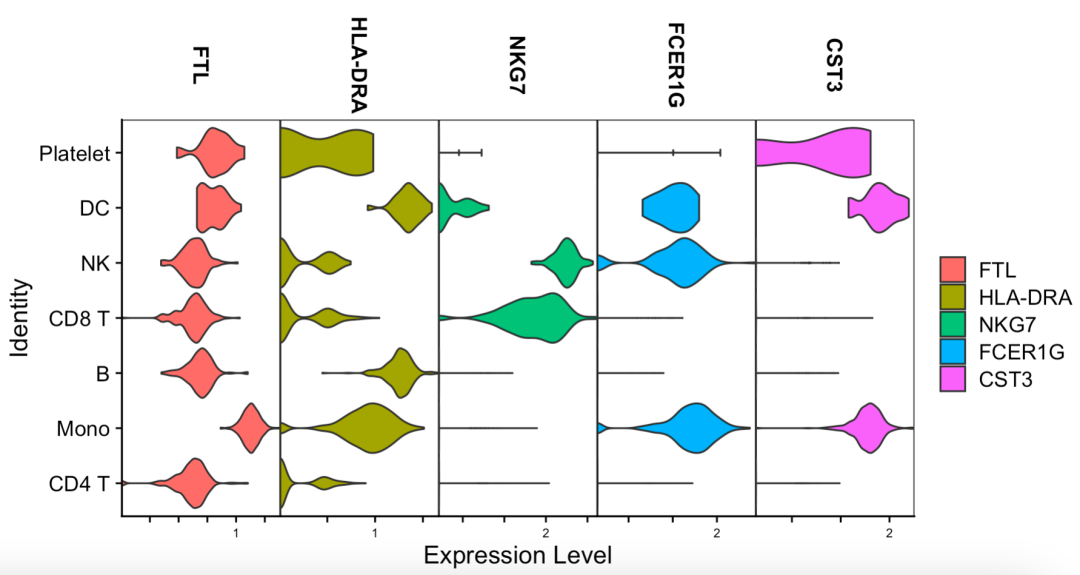
决策树模型的5个基因对照前面的决策树模型看:
- 然后是HLA-DRA可以区分B细胞以及树突细胞和其它细胞,其中B细胞以及树突细胞的区分靠CST3
- 然后T细胞里面的CD4靠NKG7区分出来,然后CD8和NK细胞靠FCER1G区分
这样的模型就非常容易解释清楚,比起前面的随机森林,LASSO回归,支持向量机来说。









