遗传效应解释了从基因突变到复杂疾病发展的因果关系。因此,全面识别遗传效应可以为疾病的发展和治疗提供有价值的见解。但如何解释复杂表型的遗传因果关系一直以来都是学界悬而未决的热点问题。近日,清华大学脑与认知科学研究院戴琼海院士团队在Cell Press旗下Patterns期刊发表了一篇题为“Explaining the Genetic Causality for Complex Phenotype via Deep Association Kernel Learning”的文章,提出可以通过深入关联核学习(DAK)解释复杂表型的遗传因果关系。
Cell Press细胞出版社特别邀请论文通讯作者之一CAAI理事长、清华大学戴琼海院士接受采访,介绍了该研究工作开展的背景、亮点以及下一步的研究计划。点击“阅读原文”或识别下图二维码阅读英文原文。
1. 利用深度学习从基因组推断复杂的因果信号
2. 在不同类型的因果变异上验证模型
3. 通过对框架的可解释分析来解释模型的原理
4. 将该模型应用于癌症、认知疾病等真实数据集
论文简介
基因突变以许多不同的方式引起复杂的疾病,遗传效应解释了从基因突变到复杂疾病发展的因果关系。因此,全面识别遗传效应可以为疾病的发展和治疗提供有价值的见解。全基因组关联分析(GWAS)被广泛用于从复杂的生物表型中发现潜在的致病位点。经典的GWAS模型假设单个位点独立地影响疾病,风险随低频基因位点的数量线性增加,因为只能发现具有强关联和直接关联的基因变异。基于基因通路的方法能够同时考虑一组具有生物意义的基因。例如,基因集富集方法通过融合单基因位点的统计值来获得通路整体的统计分数,其变体方法使用核回归(SKAT)进行关联测试。
然而,现有的全基因组关联分析(GWAS)方法大多数建立在线性假设和简单的疾病模型下,依赖于一些预先假定的遗传模型来进行人工基因型编码。而在现实实践中,复杂疾病的基因遗传效应是未知的。因此,需要一种能够合理地推断基因型与表型之间内在关系的GWAS模型。
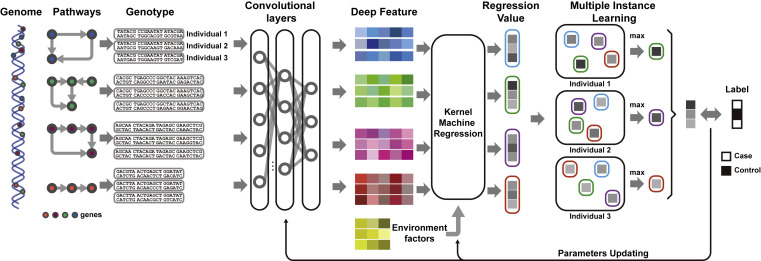
此项研究引入了深度关联核学习(DAK)模型,一种在深度学习框架中构建的GWAS方法,它可以在基因通路水平上实现自动基因型编码。在基因研究领域,虽然深度学习已经在基因调控、突变效应预测和结合位点识别方面取得了成功,但尚未建立解决一般GWAS问题的深度学习模型。在这项研究中,DAK框架结合卷积网络来编码原始SNP,提取遗传隐变量表示;核回归方法对编码的遗传表示进行进一步统计推断,预测疾病状态。更重要的是,这个内核回归层允许对自动推断的遗传表示进行统计检测,揭示疾病相关的生物过程。卷积层和核回归层都是以端到端的方式使用多实例损失函数联合训练的。因此,DAK不依赖于预先假定的遗传模型,以纯数据驱动的方式学习所有模型参数。
这项研究将DAK与七种具有代表性的基于基因/路径的方法进行了比较:经典统计方法(Burden test)、富集方法(GATES、HYST和SPU)和核方法(SKAT和SKAT-o)。DAK在包括加性、乘性、显性、隐性和杂合效应在内的广泛遗传模型下表现出了较好的性能。
研究进一步将DAK应用于四个采集的疾病数据集,包括胃癌(GC)、结直肠癌(CRC)、肺癌(LC)和精神障碍疾病,DAK模型发现了潜在的致病通路与生物解释,如检测出心肌疾病通路与精神分裂症之间的隐藏关系。
作者指出,DAK模型兼具生物学贡献和计算贡献。它不仅能够理解非线性、复杂的遗传因果关系,并改进对疾病的功能研究,还将核学习和关联分析结合在一个可解释的深度学习框架中。
Cell Press细胞出版社特别邀请戴琼海院士团队进行了专访,请他们为大家进一步详细解读。
CellPress:
您能简单介绍一下什么是深度关联核学习(Deep Association Kernel Learning, DAK)?它与全基因组关联分析(genome-wide association study, GWAS)有何关联?
戴琼海院士:
深度关联核学习(DAK)是通过深度网络特征变换,强化核方法(kernel method)表征能力,实现多尺度特征构建与关联的深度模型。包含了两个主要功能,一是在非规则化、复杂数据空间中实现规则化的特征学习;二是在特征与样本表型之间构建关联分析框架。
全基因组关联分析GWAS,是通过大量样本,寻找复杂疾病与遗传因素关联的研究方法。科学家运用GWAS发现了许多重要疾病的易感基因位点,为药物研发、疾病治疗做出了贡献。但随着研究的深入,大家发现GWAS对基因与疾病是固定关系的假设是不成立的,基因对于疾病作用过程是复杂的、非特异的,受到人种、环境等诸多因素的影响。
我们通过DAK,建立了基因与疾病关系的模型,实现了对于复杂、非线性、未知基因致病方式的自主统计推断。
CellPress:
您和您的团队引入了DAK模型,实现了对基因间复杂关联的检测,这将在遗传学、医学等领域带来哪些影响?
戴琼海院士:
在遗传学与医学领域,目前对于复杂疾病(如癌症、神经退行性疾病等)的致病机理了解仍然是非常有限的。主要原因是,目前已知的疾病易感基因位点,只是致病风险的一部分,而非疾病的本质。DAK模型可以在方法上,将未知但是显著的致病基因发掘出来,为疾病提供更多样的遗传学解释。同时DAK方法将基因以生物功能通路为单位进行分析,而生物通路在医学研究中往往具有明确的功能含义,因而有助于医学中对疾病机理的探索。
CellPress:
您能谈一谈核学习的未来吗?在该项研究中它被应用于改进全基因组关联分析,下一步将会是什么?
戴琼海院士:
核学习是一种高效的样本度量学习方法,正在被广泛应用于生物机理解析的方方面面。在未来的发展方向中,有一个重要角度是从统计意义上诠释相似样本产生的原因,增强核学习的可解释性,系统的解释一系列复杂疾病的致病机理,这对于拓展其在不同学科应用具有重要意义。
CellPress:
基于此项成果,后续研究还有哪些工作要进行?您的团队下一步的研究方向如何
戴琼海院士:
在这项工作中,我们方法已在癌症数据与神经疾病数据上进行了初步的应用验证。我所在的团队,清华大学脑与认知科学研究院,主要围绕脑科学的一系列重大问题开展研究,包括一系列先进成像与调控技术的开发、生物智能机理的研究、脑启发的人工智能,以及一系列人工智能方法的应用等等。基于此项成果,在后续研究中,我们一方面将充分利用团队在介观生物观测仪器开发上的优势,建立一整套全链条的大规模介观数据分析与挖掘平台,包括全脑范围单细胞分辨率神经信号分析揭示一系列大脑工作机制,结合大范围三维活体观测系统实现大规模的表型筛选和生物挖掘,助力药物筛选以及高通量生物发现等等;另一方面通过与众多医院展开合作,对于发现的生物结论进行深入探索,结合大量临床数据,为一系列复杂疾病提供更多诊疗思路与治疗靶点,并且同时拓展方法的疾病应用范围。
CellPress:
该研究汇聚了来自不同国家、不同学科领域的研究人员,在研究进行过程中,团队是否遇到一些困难,又是如何解决的呢?
戴琼海院士:
我们在这项研究中与哈佛大学David C. Christiani教授、南京医科大学王美林教授团队进行合作。我的团队从事人工智能与生命科学的交叉研究,Christiani教授侧重于环境基因学与生物统计学研究,王美林教授团队关注于公共卫生科学与重大疾病研究。交叉学科要求团队能够结合众多方向的研究方法和成果,从事这样的研究经常是在黑暗中摸索,可借鉴的经验少、超出预期的困难多。从整个计算方法的设计,到方法的统计效力、到具体的实践、再到应用到特定疾病的生物发现,每个环节都极具挑战。我们的方法是,综合各学科最精英的科研人员、开展深入频繁的研讨,用严谨的方案和考究的细节,把性能优化到极致、把风险和困难降到最低,大胆设计,匠心实践,把工作扎扎实实做出来。我始终认为在做科研的过程中,要时刻注意理学思维,工科实践,做问题驱动下的原始创新,要勇于拥抱交叉学科,交叉学科的思维养成后将使我们终生受益。
戴琼海,CAAI理事长,清华大学自动化系教授,清华大学生命科学学院兼职教授,清华大学信息学院院长,清华大学脑与认知科学研究院院长,中国工程院院士,中国人工智能学会理事长。近年来,主持研制了全球视场最大、数据通量最高的多维多尺度高分辨率计算摄像国家重大科研仪器,在清醒动物全脑皮层神经元活动和连接观测等领域取得了系列突破,构建了新型介观脑观测技术与仪器体系,支撑脑科学与医学的新发现,为人工智能发展提供了新途径,相关成果先后发表于Nature Photonics, Nature Methods, Nature Machine Intelligence, Patterns等期刊上。
王美林,男,博士,2010年获南京医科大学公共卫生与预防医学博士学位,2013-2014年分别在美国Wake Forest大学和Duke大学从事博士后和访问学者工作。2010年起任讲师,先后破格任副教授、教授。现任南京医科大学生殖医学国家重点实验室PI、博士生导师。2015年获江苏省杰出青年基金,2018年获国家优秀青年科学基金。
主要从事生殖内分泌激素与肿瘤生成作用研究,围绕基因遗传变异,结合表观遗传学,研究激素在肿瘤发生和发展中的调控机制。近年来研究成果先后发表在Nat Genet、Nat Commun和Cancer Res等期刊上。
北京航空航天大学宇航学院教授,博士生导师。近年来,围绕航天智能、医疗智能及金融智能,以第一/通讯作者身份在Nature Methods及IEEE Transactions等刊物发表论文30余篇,其中影响因子高于10的期刊论文8篇,并在Springer出版人工智能英文专著一部。曾获得IEEE Transactions on Fuzzy Systems 2020年度最佳论文奖(第一作者),Microsoft Research Fellow,中国自动化学会、中国人工智能学会优秀博士论文奖 担任AAAI、IJCAI及ICCV等多个人工智能会议的程序委员。
研究成果发表于在Cell Press旗下Patterns期刊上
点击“阅读原文”或扫描下方二维码查看论文
Explaining the Genetic Causality for Complex Phenotype via Deep Association Kernel Learning
https://www.cell.com/patterns/fulltext/S2666-3899(20)30068-4
https://doi.org/10.1016/j.patter.2020.100057











