文章简介
近日,厦门大学与嘉庚创新实验室利用机器学习联用方法,在计算电解液动态核磁共振(NMR)谱解析领域取得重要突破,实现了对双氟磺酰亚胺锂(LiFSI)/二甲醚(DME)电解液中动态的⁷Li核磁共振化学位移的预测。预测结果精准揭示了⁷Li核磁共振化学位移的反转现象,与实验观测结果吻合。该成果是团队继电池正极材料动态核磁谱研究和NMRNet深度学习框架之后,在相关领域的又一次方法迭代与体系应用拓展,相关研究成果已发表于顶级化学期刊《Journal of the American Chemical Society》。
该项研究成果的通讯作者为厦门大学程俊教授和汤富杰副教授,厦门大学化学化工学院2023级博士研究生尤祺和博士后孙岩为共同第一作者,嘉庚创新实验室副研究员王锋在研究过程中提供了协助。该研究工作受到国家自然科学基金(资助号:22021001、22225302、21991151、21991150、92161113)、中央高校基础研究基金(资助号:20720220009)和国家重点研发计划(资助号:2024YFA1210804)的资金支持。
研究背景
核磁共振(NMR)谱作为一种无损且对局域结构敏感的表征手段,适合用于溶剂化结构中特定原子核化学环境的表征。此外,实验NMR谱还能揭示弛豫时间、交换动力学等信息。然而,将观测到的NMR谱变化与内在分子结构变化联系起来是一项极具挑战性的任务。一些采用密度泛函理论(DFT)计算的研究,通过复杂的采样方法进行团簇提取,能够为电解质NMR谱的变化趋势提供有限信息。已有许多静态计算尝试利用第一性原理方法计算¹H、⁷Li、¹⁷O、²⁵Mg、⁴³Ca、⁶⁷Zn NMR谱来解释结构-光谱关系。尽管如此,实验化学位移反映的是来自不同局域位点的加权平均,融合了局域结构和动力学信息,这种统计平均使得信号分辨变得复杂,也加大了解析谱构关系的难度。
分子动力学(MD)模拟能够借助经典力场方法、第一性原理方法以及机器学习方法来捕捉各类电解质中的动态结构变化。但由于从MD模拟构型中获取单一光谱响应的计算成本高昂,且在复杂电解质体系中运用NMR-DFT计算时构型采样的选择过于复杂难以实施,因此将分子结构与实验光谱观测值直接联系起来仍颇具挑战。不过,已有研究尝试通过对固态结构的结构描述符及其相应 NMR化学位移进行训练,使机器学习模型在保持高精度的同时提高化学位移预测速度。目前,关于电解质动态结构特征与其实验光谱观测值之间的关系尚未达成明确共识,因此一种强大的计算方法至关重要。此外,验证模拟结果并将其与实验观测结果相关联极具挑战,但却是衡量模拟可靠性的重要基准。
研究内容
![]()
![]()
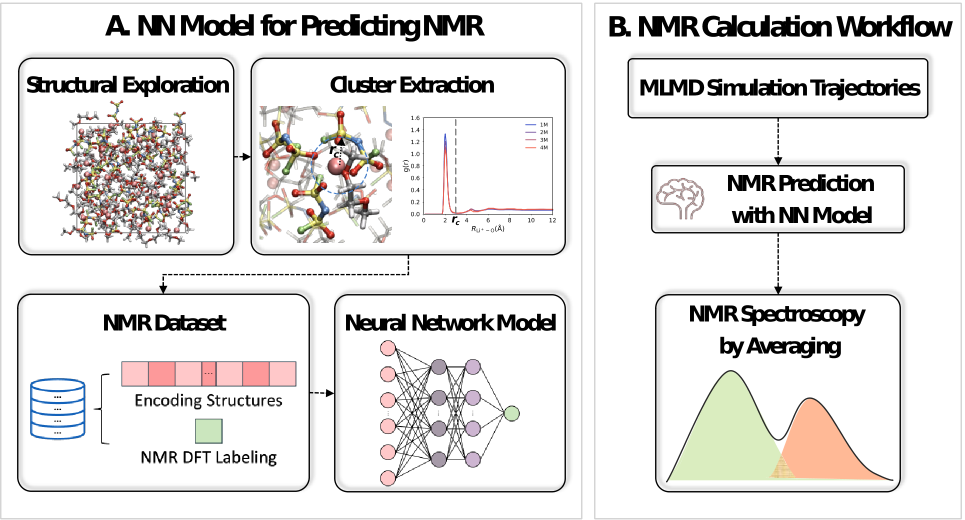
图1: 预测核磁共振(NMR)谱的流程。(左)训练神经网络(NN)模型的方法。捕捉不同浓度下的各种结构,并提取Li⁺的溶剂化结构。随后,使用描述符对结构进行编码,并计算其相应的化学位移。(右)NMR谱预测流程。使用机器学习分子动力学(MLMD)模拟生成轨迹,然后利用所得到的核磁共振预测神经网络模型来获取核磁共振光谱。
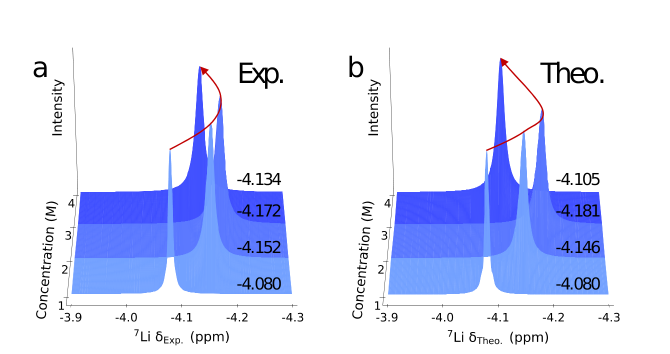
预测核磁共振(NMR)谱的流程如图1所示。构建神经网络(NN)模型时,我们先从MLMD轨迹中稀疏采样构型,提取锂离子周围的第一溶剂化层,将其作为团簇并加以标记,并运用LMBTR描述符对结构进行编码,进而得到一个基于密度泛函理论(DFT)的数据集,其中包含约28000个⁷Li化学位移数据。随后,我们利用LiFSI/DME溶液对基于神经网络的核磁共振谱模型展开验证,测试集中⁷Li各向同性值的均方根误差约为0.13 ppm。获得神经网络模型后,我们对四个浓度下LiFSI/DME溶液的机器学习分子动力学模拟得到的轨迹预测核磁共振谱。我们对四个浓度下的轨迹进行等间隔抽取,确保每种浓度下锂离子数量约为90000个,随后由LMBTR描述符编码Li⁺的溶剂化结构,作为神经网络模型的输入来预测核磁共振化学位移,预测结果如图2b所示,其与实验结果(图2a)在不同浓度电解液中的变化趋势相似,在4 M浓度时均出现了⁷Li化学位移的反转现象。
![]()
![]()
图2: NMR谱预测结果与实验结果的对比。1 M到4 M LiFSI/DME溶液的(a) 实验NMR谱。(b) 神经网络预测的NMR谱。红色曲线用于直观展示NMR谱随着浓度增加的变化趋势。
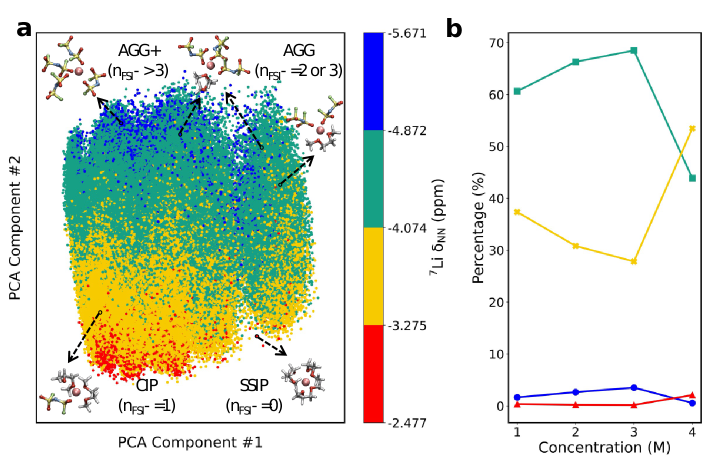
为揭示谱构关系,深入解析因浓度变化导致NMR谱位移变化背后内在的分子结构演变机制和相互作用规律,我们采用了无监督主成分分析(PCA)对锂离子的局域结构的描述符进行降维处理,结果如图3a所示。我们发现主成分PC#1反映了溶剂化结构的对称性以及Li⁺周围分子的取向变化;而主成分PC#2则捕捉到了Li⁺周围的局域环境信息。化学位移值最大的区域(红色)与溶剂分离离子对(SSIPs,此时溶剂壳层内的FSI⁻数量nFSI⁻=0)相关;黄色区域对应接触离子对(CIPs,nFSI⁻=1);绿色区域与聚集体(AGGs,nFSI⁻=2或nFSI⁻=3)相关联;而化学位移值最小的情况,出现在AGGs和AGGs+中,此时nFSI⁻≥3。这一发现符合化学直觉,也展现了主成分分析方法能够有效地捕捉局部环境变化对周围电子密度的影响。图3b揭示了浓度从3 M到4 M时,占主导的中等(黄色)和较高(绿色)NMR值(绝对值)的结构,前者占比上升,后者比例下降。推测CIP(黄色)结构占比上升,是因Li⁺-Li⁺相互作用促使高度局域化的AGGs+结构出现。
![]()
![]()
图3: (a) 对不同浓度下锂离子Li⁺溶剂化结构模式的主成分分析(PCA)。每个点的颜色编码代表相应的核磁共振化学位移值。x轴和y轴表示两个最重要的主成分PC#1和PC#2。代表性片段的颜色标记如下:红色代表溶剂分离离子对(SSIP),黄色代表接触离子对(CIP),绿色代表聚集体(AGGs),蓝色代表FSI⁻阴离子数量nFSI⁻=3的AGG以及nFSI⁻>3的AGGs+。(b) 不同颜色的代表性溶剂化结构比例随LiFSI浓度的变化情况。
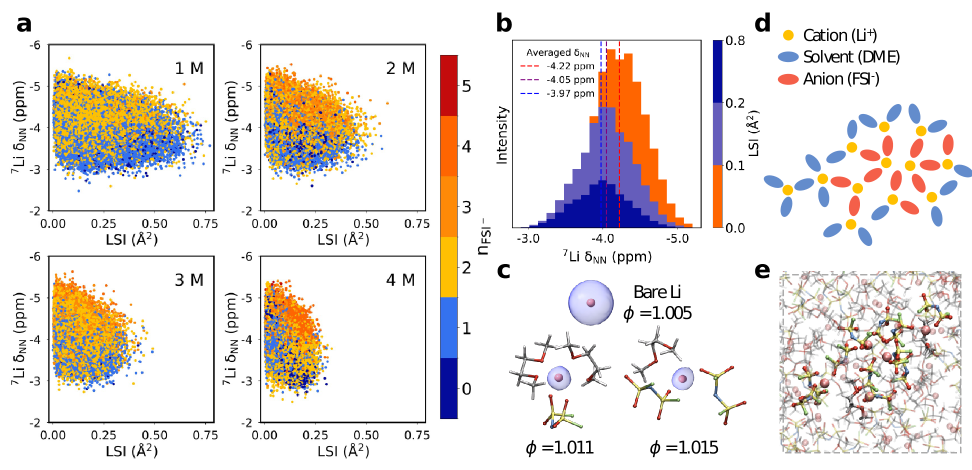
进一步来看,主成分分析(PCA)的结果得到了nFSI⁻和LSI(局域结构指数,表征Li⁺周围的氧原子径向距离分布的不均匀性)等局域结构参数的支持。图4a展示了核磁共振化学位移值随各种LSI值的变化情况,颜色根据nFSI⁻来区分。处于较高nFSI⁻溶剂化环境中的Li⁺,其化学位移往往向高场方向移动,反之亦然,这为PCA中溶剂化结构的区域划分提供了依据。此外,我们将LSI划分为三个范围,并计算了相应的平均化学位移,其直方图见图4b,这表明较小的LSI范围主要对应较低的化学位移范围。随着化学位移值减小,LSI值降低,这种趋势在四种浓度下一致。造成这一现象的原因在于,当配位数达到较高水平nFSI⁻≥4的构型倾向于形成填满亚间隙结构的长链簇集。与此同时,在4 M浓度下,配位数为nFSI⁻=0或1的溶剂化结构(蓝色区域)伴随着nFSI⁻≥4的溶剂化结构出现,如4d与e所示,这些结构围绕着AGGs+类型的聚集体分布,从而使整体LSI值相较于3 M浓度更低。此外,我们对1 M至4 M浓度范围内的一些溶剂化结构进行研究,以量化锂原子核的电子定域函数(ELF)的形变因子ɸ,1-4 M浓度下的平均值分别约为1.0130±0.0002、1.0128±0.0002、1.0127±0.0001和1.0130±0.0001,图4c展示了ELF形变程度变化的示意图。如图所示,随着形变因子增加,ELF形变更加明显。当变形更显著时,锂原子核周围的电子屏蔽减弱,导致化学位移向低场移动。因此,平均形变因子从1 M到3 M下降,从3 M到4 M上升,这与核磁共振化学位移先向高场移动、再向低场移动的情况一致。
![]()
![]()
图4: (a) 每种浓度下局域结构指数(LSI)与化学位移值之间的相关图。各点的颜色与nFSI⁻的值相匹配。(b) 不同LSI范围对应的化学位移直方图,红色、紫色和蓝色虚线分别表示对应于LSI范围为0.1 Ų、0.1~0.2 Ų和0.2~0.8 Ų的平均化学位移。(c) 围绕锂原子核(粉红色球体)具有不同形变因子ɸ的电子局域函数(蓝色)的示意图。(d), (e) Li⁺与FSI⁻长链团簇的示意图,元素的颜色标记如下:锂(Li)为粉红色,碳(C)为灰色,氢(H)为白色,氧(O)为红色,硫(S)为黄色,氮(N)为蓝色,氟(F)为绿色。
总结与展望
该项工作提出一种基于机器学习的方法,联用MLP和NN模型,计算 LiFSI/DME溶液的动态NMR谱。LiFSI浓度从1 M增至3 M时,溶剂化结构变化使NMR化学位移向高场移动,4 M时则向低场移动。我们构建了分子结构与NMR谱的定量关系,深入剖析了溶剂化结构归属。研究表明,存在两种相互竞争的局部溶剂化结构,电解质浓度接近上限时,其主导地位交替,导致⁷Li化学位移变化。该方法精准高效,预测结果与实验谱高度吻合。有望拓展到其他复杂电解质体系,并预测其他原子核化学位移。
这项工作为理解溶剂化结构与NMR化学位移关系提供了新视角,是研究电解质溶液的高效手段,加深了对电解质溶剂化结构的理解,为优化电解质设计开拓了新路径。
致谢
感谢华东师范大学胡炳文教授和上海科技大学刘海铭教授的宝贵建议。程俊教授感谢国家自然科学基金(资助号:22021001、22225302、21991151、21991150、92161113)、中央高校基础研究基金(资助号:20720220009)、人工智能应用电化学实验室(AI4EC)、IKKEM(资助号:RD2023100101和 RD2022070501)的资金支持。汤富杰副教授感谢科技部重点研发计划(资助号:2024YFA1210804)和厦门大学的启动资金支持。
论文链接:
https://pubs.acs.org/doi/10.1021/jacs.5c02710
纳米人学术QQ交流群
(加群方式:请备注:姓名-单位-研究方向(无备注请恕不通过),由管理员审核后入群。)一维材料群-1:463352826
二维材料群-2:1075914107
复合材料群-2:834770793
可穿戴器件学术群:1032109706
光催化群-2:927909706
电池讨论群-1:418038617










