RNA可变剪接(Alternative splicing)是基因转录后一种重要的调控机制,也是生物体多样性和蛋白质多功能性的重要来源之一。人类约90%以上的基因存在可变剪接,不同组织与细胞类型中可变剪接的多元性促进了细胞表型的多样性。同时,引起RNA可变剪接的变异也与人类多种遗传疾病相关。 值得注意的是,RNA可变剪接具有组织特异性,相同的pre-mRNA序列能以组织特异性的形式发生可变剪接,从而产生多样性的转录组和蛋白质组表达。然而,现有算法无法预测组织特异性的可变剪接,因此亟需开发能够精准预测组织特异性可变剪接的算法工具,加深我们对于遗传变异的解读及后续研究。
近日,浙江大学良渚实验室沈宁/刘志红课题组在Nature Communications上发表了题为“SpliceTransformer predicts tissue-specific splicing linked to human diseases”的研究论文,开发了基于Transformer架构的多模态深度学习模型SpliceTransformer(简称SpTransformer),用于预测pre-mRNA序列中的组织特异性可变剪接位点。SpTransformer可以用于解析组织特异性剪接变异相关的疾病,为疾病相关遗传变异提供基于可变剪接机制的全新见解。
文章发表在Nature Communications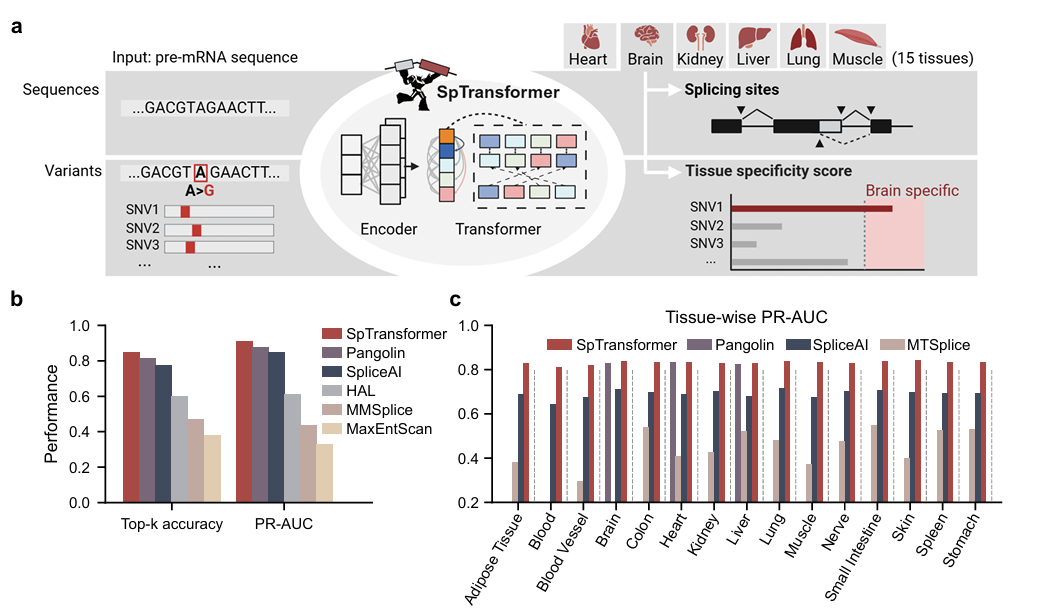
图1. SpTransformer模型仅以序列为输入,预测15种人体组织中的组织特异性剪接。该模型可用于评估遗传变异并预测组织特异性的剪接变化,其性能明显优于其他已有算法
SpTransformer在训练数据和算法架构上均有所创新。SpTransformer基于GTEx人体组织RNA-seq数据和额外的哺乳动物(恒河猴、小鼠、大鼠)组织的RNA-seq数据训练,从多个数据集中学习可变剪接相关的序列特征。模型使用one-hot编码的pre-mRNA序列作为输入。序列经过卷积编码器处理后,通过一个8层包含自注意力模块的Transformer网络,输出多标签分类结果。该方法基于9000nt~15000nt的长序列上下文信息,对输入序列中央的1000nt长度序列同时做预测,既能预测出序列中存在的可变剪接位点,也能为每个位点进行多标签分类,预测位点在15个主要人体组织中是否会被使用到。为了考虑尽可能长的序列上下文信息,该模型结合了卷积编码和Sparse Sinkhorn Attention稀疏注意力算法,鼓励模型考虑远端序列之间以片段为单位的互作效果,而避免逐一计算碱基和碱基之间的长距离互作关系,这样的做法允许模型以线性复杂度处理大量数据,从而规避了传统Transformer模型处理超长序列时无法承受显存开销的问题。最终,该模型对可变剪接的预测结果明显优于已有算法,且创新地实现了对组织特异性剪接的预测(图1)。
之后,研究团队针对模型的可解释性进行了探索。研究团队通过可视化分析模型考虑不同序列元件的权重,发现SpTransformer模型可以成功发现1000b p以外的远端序列调控元件对可变剪接的影响。同时,模型在预测不同组织的可变剪接时,会考虑不同的序列motif,其中既有已知的经典序列调控元件,也包含未被研究过的de novo motif。
图2. SpTransformer模型可用于评估突变对剪接的影响
随后,研究团队应用SpTransformer预测单核苷酸突变(single nucleotide variant,SNV)对组织特异性剪接的影响。通过分别预测突变前序列和突变后序列的剪接情况,并计算它们预测分数的差异,模型以数学方法将突变对目标区域可变剪接的影响量化为ΔSplice分数(图2)。研究团队批量预测了大型数据库ClinVar中收录的1,273,053个SNV,发现突变的致病性和影响可变剪接的情况有较强的关联。在非编码区域突变中,通过SpTransformer给出的ΔSplice分数,能以超过0.98的ROC-AUC区分致病(Pathogenic)和良性(Benign)突变。同时,模型将大量原本标注为效果未知(Uncertain significance)的突变注释为影响可变剪接,一定程度上填补了突变效应注释的空白。
图3. SpTransformer模型可用于评估突变对组织特异性剪接的影响,并从大规模数据分析中识别可能影响组织特异性剪接的突变及其富集的基因
尽管可变剪接的组织特异性早已为人所知,但突变如何影响这种特异性剪接仍然是个未充分探索的领域。为评估单核苷酸变异(SNV)对组织特异性剪接的影响,研究团队开发了Tissue z-score分数,用以衡量突变对特定组织中剪接模式的影响是否显著高于其他组织。通过分析GTEx RNA-seq数据中的非组织特异性剪接位点,并模拟这些位点附近发生随机突变,研究团队建立了用于参考的统计学分布。当待预测SNV的Tissue z-score明显大于参考分布中的值时,就认为该SNV具有组织特异性。利用这种方法,研究团队从ClinVar数据库中识别出可能影响组织特异性剪接的突变及其富集的基因(图3)。研究结果显示,这些基因多与相关组织的遗传疾病有关,但不一定表现出组织特异性的表达模式。图4. 算法针对三种精神疾病数据展开分析,从组织特异性剪接改变角度解读精神疾病发生的潜在机制
为了深入探讨SpTransformer在疾病诊断和治疗中的应用潜力,研究团队利用该算法分析了与自闭症(Autism),精神分裂症(Schizophrenia)和双相精神障碍(Bipolar disorder)相关的超过17万个样本的全外显子组测序结果。这些样本涵盖了患者、患者家庭成员和健康对照组。从超过千万的未知突变中,SpTransformer筛选出大量可能影响可变剪接的突变。
研究团队深入分析了这些影响剪接的突变,发现脑组织特异性的剪接改变在三种类型的精神疾病中均有显著富集。进一步的基因表达量分析揭示,这些突变所在的基因,不仅包括在大脑中特异性表达的,也包含在多数组织中广泛表达的基因,表现出双峰分布的特点。GO富集分析(Gene Ontology enrichment analysis)显示,由模型筛选出的基因与脑组织功能存在紧密联系(图4),在脑组织中特异性表达的基因通常与突触信号传导通路相关,而非组织特异性表达的基因则富集在细胞骨架相关通路。这一发现进一步揭示了脑组织中特异性剪接和特异性表达之间存在相对独立性,即使是在多种组织中普遍表达的基因,也可能通过剪接变异对脑组织产生重要影响,进而可能引发相关疾病。
同时,虽然从这三种精神疾病中筛选出的基因富集到了某些相同的通路,但每种疾病有其独特的致病基因及突变。大规模文献搜索结果显示,由SpTransformer识别的许多基因已有相关文献支持其与特定疾病的关联,证明了该工作预测的准确性。此外,还有较多新发现的基因,目前尚未有相关研究报道,这些基因可能为未来的精神疾病研究提供新的线索和方向。
此外,研究团队也针对肾脏特异性剪接进行了进一步的数据分析。模型在糖尿病肾病相关数据上进行实战,经由RNA-seq方法进行验证,以83%的准确率预测出了影响肾脏中可变剪接的突变。
以上结果表明,SpTransformer从组织特异性可变剪接的角度出发,有潜力发现传统基因表达量分析无法找到的疾病成因。这为理解疾病背后的遗传因素提供了除基因表达水平以外的重要视角。进一步地,基于组织特异性的可变剪接分析,有望成为解析复杂疾病遗传机制的关键方法之一。
图5. 组织特异性可变剪接分析算法SpTransformer的概念图
综上,该研究开发了一个精准地预测具有组织特异性的可变剪接的算法工具SpTransformer,并通过大量真实突变数据,验证了其在遗传诊断中预测组织特异性影响RNA可变剪接的致病突变的能力,具有重要临床价值和研究意义。文章相关代码已开源,发布在GitHub平台(https://github.com/ShenLab-Genomics/SpliceTransformer)。此外,研究团队还提供了一个便捷的在线服务平台(http://tools.shenlab-genomics.org/tools/SpTransformer),使用户能利用SpTransformer快速预测突变对组织特异性剪接的影响。
浙江大学良渚实验室沈宁研究员和刘志红院士为该论文的共同通讯作者,交叉培养博士生游宁远为本文的第一作者。浙江大学管敏鑫研究员、裴善赡研究员、南京大学蒋松博士、施劲松博士、复旦大学孙思琦研究员协助监督指导了本项工作,良渚实验室多位研究员及沈宁课题组多名成员对该工作作出了重要贡献。
良渚实验室沈宁课题组围绕“组学与精准医学分析算法开发与应用”开展临床转化密切相关的研究,运用生物信息学数据整合分析与人工智能算法,并结合实验筛选平台进行药物研发与精准治疗。
课题组目前有多项具有重要应用价值的课题正在推进,与著名医学专家主导的实验室有合作关系,诚招具有实验生物、计算生物背景的博士后和研究助理。
详细招聘信息见:
https://person.zju.edu.cn/shenning
shenningzju@zju.edu.cn
原文链接:
https://www.nature.com/articles/s41467-024-53088-6·END·

快点亮"在看”吧










