

在化工领域中,乙烯环氧化的反应问题与数学领域著名的哥德巴赫猜想类似,是所在领域公认的、存在已久的难题。
乙烯环氧化是乙烯化工领域中重要的反应之一,其发展历史至今约 50 年,目前国际上已能够达到千万吨级别的工业生产规模。
虽然该方向的理论研究和实验已有多年的研究经验,但领域内对于乙烯环氧化的反应机理尚未形成统一的共识,仍有许多基础性的物理化学问题存在争议。
例如,银催化剂中的催化位点,是氧化银还是金属银更具有催化活性?
究其原因,一方面,理论研究的主要问题在于,研究人员只能通过基于量子力学的第一性原理进行计算,一些结构在被拆解后“似是而非”。
另一方面,在实验科学中,银催化剂表面结构通常在高温、高压条件下产生,科学家们通常使用离线检测的方法。
但这种手段仅可获得相对模糊的振动谱学信息,很难在这种反应条件下探测到活性位点,因此对反应过程中的具体机理不甚明晰。
基于此,复旦大学刘智攀教授团队利用机器学习原子模拟,揭示出银催化乙烯环氧化活性位点。
在该研究中,确定了银(Ag)(100)表面对乙烯环氧化具有选择性的原因:在反应条件下形成的 Ag(100)表面的复杂重构中,存在五个亚表面配位氧原子(O5)。该研究不仅进行了创新的理论计算,还用实验加以证明,为领域中银催化乙烯环氧化反应的活性位点的长期争论问题提供答案,有利于推动研究人员未来在原子级别设计与调控催化剂。
审稿人评价称,该研究是通过机器学习辅助的第一性原理计算和统计力学计算的完美结合。另一位审稿人认为,该论文在技术上非常先进、全面和系统,并且结果具有非常重要的工业意义。

图丨刘智攀(来源:刘智攀)
近日,相关论文以《方形锥体次表层氧[Ag4OAg]驱动银的选择性乙烯环氧化反应》(Square-pyramidal Subsurface Oxygen [Ag4OAg] Drives Selective Ethene Epoxidation on Silver)为题发表在 Nature Catalysis 上[1]。
复旦大学博士研究生陈东晓和陈林为第一作者,刘智攀教授担任通讯作者。

图丨相关论文(来源:Nature Catalysis)

捅破技术的“窗户纸”,实现 1 周内完成结构空间采样 50 万个
化学反应的复杂性在于,反应机理、反应的路径以及表面结构的未知性。
在该研究中,机器学习原子模拟可以真正深入到催化的原子层级,其背后离不开“第一工具”——机器学习。
但一般的机器学习势函数的问题在于,没有办法进行足够复杂的势能面结构采样,因此预测能力相对有限。
2022 年,随着 AI 技术的发展和该团队多年的技术累积,他们意识到,捅破探索银催化乙烯环氧化活性位点这层“窗户纸”的时机到了。
该研究历时约两年的时间,研究人员基于机器学习方法,从理论上对银催化剂表面结构进行类似“数字孪生”的演示。
首次揭示出清晰的表征图像,包括从反应条件下的银催化剂表面 Ag(100)、Ag(111)、Ag(110)重构,再到乙烯环氧化。
刘智攀表示:“我们通过机器学习原子模拟,能够提供底层的所有可能性和所有数据,有了这些数据我们就可以将量子力学计算、微观动学模拟、在线的实验设备组合起来,把乙烯环氧化机理的历史问题弄清楚。”
研究人员对不同构型的 Ag 表面结构进行筛选后,确认 3 个 Ag 表面在 500K、1bar 乙烯,1bar 氧气条件下的最优表面结构。在此条件下,共确定 4 个共存相。
与其他 3 个共存相只存在表层氧不同的是,在 Ag(100)中,独特表面氧化物相 O5 相除了具有表层氧以外,还包括五配位的次表层氧[Ag4OAg],以及包含特殊电子性质的强吸附乙烯。
之后,研究人员通过自主开发的反应路径自动采样算法,对乙烯在上述表面的反应机理进行深入研究。
结果显示,仅 O5 相能够促进环氧乙烷的形成;而在其他相中,皆是先将燃烧反应至二氧化碳,对环氧乙烷选择性产生不利影响。
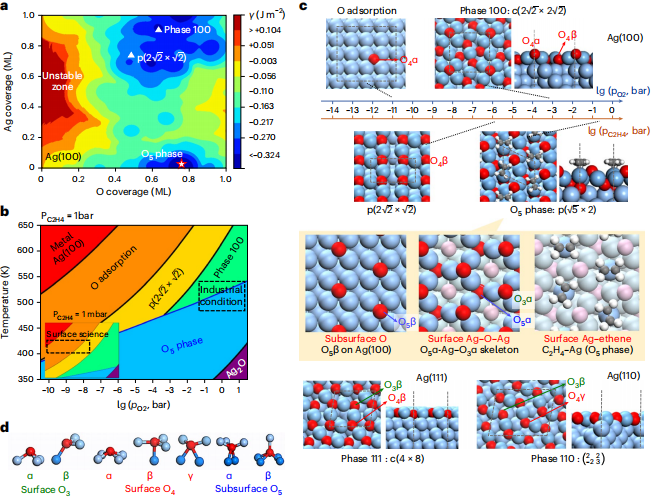
图丨最优表面相自动搜索算法获取银在反应条件下的最优表面结构(来源:Nature Catalysis)
该研究主要分为三个阶段,包括:势函数、时间成本和计算效率的评估和实验探测。
第一,势函数。势函数能够全面、准确地描述银/氧化银与碳氧氢有机化合物(例如乙烯、环氧乙烷、丙烷等)的相互作用。
据了解,此前课题组已得到银碳氢的势函数,而研究人员的目标是把它优化到 3-4 毫电子伏特每原子。只有精度达到足够高,才有可能通过该参数预测未知的结构、过渡态、反应速率等。
第二,时间成本和计算效率的评估。在该研究中,研究人员实现了 1 周内完成结构空间采样数约 50 万个。
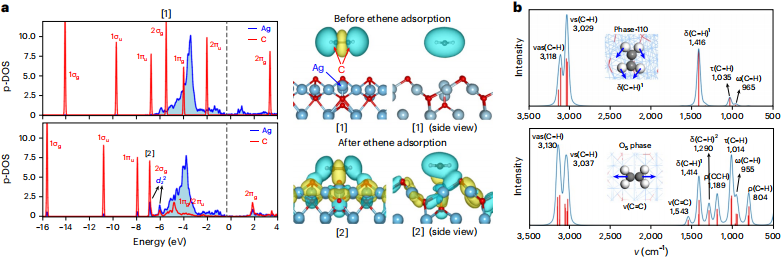
图丨O5 相上强吸附乙烯的电子结构分析(来源:Nature Catalysis)
第三,实验探测。完成理论研究后,研究人员在原位红外光谱表征后,得出乙烯吸附在磷表面引起碳-碳双键振动峰达 1583cm-1 的结果。
刘智攀回忆说道:“让人兴奋的是,这种稳定的结构在化学领域中从来没有见过。并且,该实验对应理论上对 O5 相中乙烯的红外模拟结果,证实理论对 O5 相活性位点预测的准确性。”

让催化理论计算在实验之前“跑起来”
该研究首次用机器学习的方法,通过计算揭示出乙烯环氧化反应条件下银的催化性质,为催化领域的研究打开了想象的空间。
“原则上任何催化反应、化学反应的过程,都可以通过该方法,按同样的工作流程,确定其反应条件下的表面结构。后续,还可以尝试将银替换为金、钙钛矿等,以进行其他环氧反应的验证和发现。”刘智攀表示。
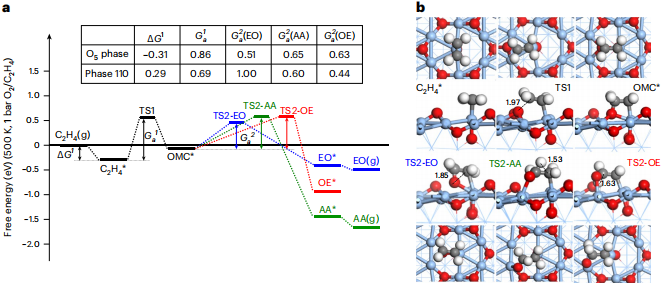
图丨反应条件下各个共存相的乙烯环氧化反应机理计算(来源:Nature Catalysis)
在科学领域,很多看似复杂的问题其实本质上很简单。
刘智攀指出,在没开发出这种新技术之前,看问题类似于“盲人摸象”,但有了这种足够好用的方法和技术,相当于站在全局的角度去看问题,所以问题也变得相对简单。
他认为,未来的科研发展趋势将由理论计算驱动主导,一方面,这种方式的成本较低;另一方面,也可以节省做实验的人力成本。
“理论计算驱动方法将改变我们的科研范式,基于此的科学研究中,包括实验室氛围、实验室架构、博士导师需要的知识储备等各方面,都将发生变化。”
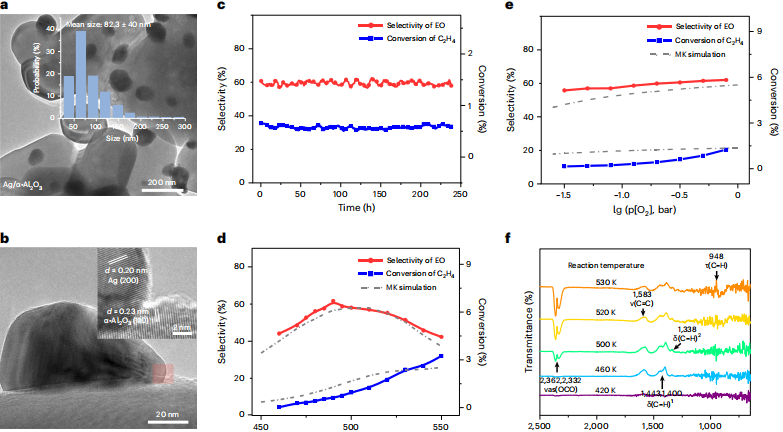
图丨银催化剂的合成、反应动力学测试、原位红外光谱测试(来源:Nature Catalysis)
此外,在化学的反应以及催化剂的设计方面,刘智攀认为,在未来 10 年内,可能会涌现出一批由理论主导,再从实验的角度认识和发现的新型催化剂。

用原始创新构筑独有的科研生态圈
刘智攀本科和硕士毕业于上海交通大学化学系,之后他在英国贝尔法斯特女王大学获得理论化学博士学位,并在英国剑桥大学从事博士后研究工作。
2005 年 8 月开始,他在复旦大学化学系成立独立课题组并担任教授,博士生导师。目前,刘智攀课题组主要研究方向是发展用于催化机理研究以及材料设计的理论计算新方法,来指导催化实验。
“我刚回国任教的时候,计算机不仅价格贵,而且计算能力十分有限。因此,那时基本领域内都是通过第一性原理计算,研究已知的体系和电子结构。”刘智攀回忆道。
他始终认为,在科研领域中国需要建立自己的原始创新,从而通过发明创新技术引领科学的发展。
近年来,刘智攀课题组通过一系列理论方法,从探索势能面到建立势函数,再到各种表面结构或微观动力学结合的反应路径,慢慢建立起一套独有的生态圈。
2013 年,他带领团队开发了一种全局优化的方法,名为随机势能面行走(SSW,Stochastic Surface Walking),该方法让寻找势能面的关键结构变得简单可行。
但是,SSW 受限于方法速度过慢,只能做 10 个原子左右的简单化学反应。
为解决该问题,刘智攀与课题组成员在固体相变方向进行长达三年的探索,从 0 开始自己写代码、探索神经网络,直到做出势函数,于 2017 年发表相关论文[2]。
“这个研究是我们课题组的一次重要突破,我们有信心将复杂的、高维度空间的材料空间预测出来,并有望用该方法在未来进行原子模拟。刘智攀说。
2018 年,他们自主开发大规模机器学习原子模拟(LASP,
Large-scale Atomic Simulation with neural network Potential)平台 ,用来预测结构和反应途径,截至目前已更新至 3.6.0 版本。
LASP 不仅作为课题组内部的研究工具,还通过网页(www.lasphub.com)为所有研究人员开放。
据介绍,2023 年,中国科学技术大学课题组构建新型人工碳晶体 C60,在实验之前正是基于 LASP 理论模拟计算进行相关实验,相关论文发表在 Nature[3]。
图丨刘智攀课题组开发的 LASP 网站(来源:LASP 官网)
最近,结合 GPU 计算,研究团队已经做出全生命探索的势函数,并且已经达到非常高的计算资源。
基于此,刘智攀认为,未来甚至有望跳过势函数的步骤,直接从材料设计和反应设计开始研究,这将大大加速领域的发现和发展。
与用基本规律来预测现实世界相比,AI 通过大数据学习和发现科学规律的方式,具有成本低、速度快、效率高等优势。
近期,该团队也在推进一些横向课题,例如企业希望通过与 AI 结合的、领先的理论计算,进行半导体领域结构、性能等方面关键数据的预测,包括原子图像、热力学、动力学等。
“未来,在材料设计、生物医药等领域,有望由 AI 主导课题发展。我们也将继续深化 AI 和催化、化学领域结合的方向,希望为该领域做出更多的贡献。”刘智攀最后说道。

1.Chen, D., Chen, L., Zhao, QC. et al. Square-pyramidal subsurface oxygen [Ag4OAg] drives selective ethene epoxidation on silver. Nature Catalysis (2024). https://doi.org/10.1038/s41929-024-01135-22.Si-Da Huang et al.Material discovery by combining stochastic surface walking global optimization with a neural network.Chemical Science 8, 6327-6337(2017). https://doi.org/10.1039/C7SC01459G3.Pan, F., Ni, K., Xu, T. et al. Long-range ordered porous carbons produced from C60. Nature 614, 95–101 (2023). https://doi.org/10.1038/s41586-022-05532-0运营/排版:何晨龙










