2024年2月8日,来自清华大学自动化系的汪小我团队在Synthetic and Systems Biotechnology上发表文章DIProT: A deep learning based interactive toolkit for efficient and effective Protein design。
文章介绍了交互式蛋白质设计工具包DIProT。DIProT利用非自回归深度生成模型来解决蛋白质逆折叠(Inverse Protein Folding)问题,并结合了蛋白质结构预测模型。通过这种整合,用户可以将先验知识纳入设计过程,并对设计进行评估形成一个有人类反馈的虚拟设计循环。在TS50和CATH4.2数据集上,科研团队的逆折叠模型在有效性和效率方面都表现出了很强的竞争力。
蛋白质由折叠成各种结构的氨基酸链组成,是多种生物功能的基础。预测给定蛋白质序列的结构经过科学家们的深入探索,目前已开发出一系列算法,包括基于能量函数的方法、基于协同进化的方法和基于端到端的方法。这些方法的成功凸显了计算方法在蛋白质相关领域的潜力。
蛋白质逆折叠问题是一个同样重要但更具挑战性的问题,它涉及设计尽可能符合给定蛋白质结构的氨基酸序列。这一任务有助于设计精细的突变蛋白质,是生物工程中全新蛋白质设计的核心步骤。
1. 设计过程和利用DIProT进行交互式蛋白质设计
蛋白质设计是一项复杂的任务,通常需要多次反复试验。这一过程通常从选择一个对所需功能具有高潜力的参考结构开始。然后通过突变或蛋白质设计算法生成氨基酸序列,并进行实验验证,以评估其结构是否接近参考结构及其功能。这些实验结果将指导保守区域的识别和其他区域的完善,从而为后续的迭代提高成功率。
最具挑战性的步骤之一是生成候选序列,为了解决这一难题并完成后续的整个设计循环,汪小我团队开发了DIProT工具包,它具有以下特点:
1.基于非自回归解码范式的高效逆折叠模型,实现经济高效的快速计算实验。2.用户友好的图形用户界面(GUI),集成多种算法,促进快速直观的反馈设计循环。3.利用深度生成模型,在参考结构和需要保留的可选序列图案的条件下,生成候选序列并对其进行排序。4.对生成序列的预测结构和参考结构进行详细的原子级结构比较,帮助用户过滤结构相似性低或具有其他不需要的特征的序列,并确定在后续生成轮中需要保留的序列。
重新设计噬菌体蛋白质(PDB ID:2xcj_A)的示例直观地展示了DIProT流程(图1b)。在本示例中,用户上传了2xcj_A的完整结构作为参考结构(pdb 格式),并指定“5”为要生成的设计数。需要注意的是,DIProT只将参考结构的主链原子坐标作为输入。这意味着用户可以上传完整的蛋白质结构来生成相似或改进版本,也可以上传仅包含主链信息的文件来进行新蛋白质设计。
一旦上传了参考结构并指定了设计数量,DIProT就会利用逆折叠模型生成多个与参考结构相匹配的序列。如图1b iii所示,这些设计将使用计算指标进行排序:(1) 分数,逆折叠模型提供的该序列的可能性(越高越好)。(2) RMSD,设计(ESMFold 预测)与参考结构之间的均方根偏差(越低越好)。(3) pLDDT,ESMFold 预测结构时的局部距离差异测试得分(越高表示越好)。这些指标从序列和结构两个方面对蛋白质设计进行了综合评价。
参考结构、特定设计序列及其预测结构都是可视化的,用户可以与它们进行交互。
科研团队描述了DIProT的整体推理流程。给定一个蛋白质结构,科研团队将从其主链原子的坐标中提取特征。然后,基于MPNN的解码器根据记忆库、之前解码的序列和当前的解码步骤生成蛋白质序列。解码器输出所有节点的潜在表示,然后由分类系统进行处理,以获得每个残基20个氨基酸的分布情况。如果解码步骤达到预定的最大值,科研团队就会从预测的分布中抽取设计序列。否则,科研团队会屏蔽预测置信度较低的位置,然后进入下一个解码步骤。
3.1 序列级评估
序列恢复率衡量的是在蛋白质结构不变的情况下,计算方法重现蛋白质天然氨基酸序列的能力,而复杂度评估的是模型对原生序列的预测能力。结果表明,DIProT的性能更好。
虽然序列恢复是评价设计模型的一个广泛使用的指标,但这还不足以判断其实际效果。因此,科研团队对单例和数据集进行了额外的评估。
科研团队参考人类γ-结晶素D蛋白(PDB id:1h4a_X)的结构生成了20个序列(图3a)。这表明,DIProT建议在35和39等位置保留原始氨基酸序列,在34和42等位置进行保守替换,并建议在31和38等位置进行高多样性替换。
在对保留测试集中的每个参考蛋白质结构运行推理过程后,科研团队得到了原始蛋白质中氨基酸的混淆矩阵。该矩阵可分为6组,与BLOSUM62所指示的保守替换组相对应(图3b)。这表明DIProT在模型训练过程中无需明确指令即可学习不同类型氨基酸之间的替换模式,展示了其强大的序列生成能力。
科研团队还评估了DIProT在不同采样温度下推荐不同序列的能力。图3c显示了采样温度对序列多样性和恢复的影响,证明DIProT可以生成更多样化的序列,而只需在序列恢复方面稍作权衡。
3.2 结构水平评估
由于蛋白质的功能由其结构决定,而结构又由序列决定,因此必须从结构层面对设计进行评估。科研团队主要考虑两个指标:RMSD和pLDDT。如上所述,RMSD反映了预测结构与原生结构之间的结构相似性,而pLDDT则反映了ESMFold的预测置信度。
与序列相似性评估类似,科研团队在数据集层面对DIProT进行了评估,并将其结果与随机突变进行了比较。图4显示,与具有相同序列恢复能力的随机突变序列相比,DIProT的设计具有更低的RMSD(中位数=1.31Å对16.38Å)和更高的pLDDT(中位数=84.36对 31.67)。这表明DIProT能够捕捉蛋白质序列与结构之间更深层次的关系,而不仅仅是优化序列相似性。为了排除ESMFold预测误差的影响,科研团队比较了ESMFold对原生序列和设计序列的预测结果。结果显示两者具有很高的相关性,进一步证明了DIProT能够生成与原生序列结构相似的序列。
科研团队使用DIProT重新设计了一个糖结合蛋白(PDB ID:2xr6),并使用ESMFold预测了其结构。结果表明,尽管序列恢复率较低(50.8%),但设计序列对应的结构与原生结构非常相似(RMSD=0.503Å)。尽管与某些位置(如N端的一小块区域)相对应的结构仍存在偏差,但要恢复这些部分不仅对逆折叠模型是一个挑战,对折叠预测模型也是一个挑战。事实上,ESMFold对原生序列的分析也显示出这些部分的预测结果存在一定的偏差。
3.3 时间效率
实时性对于交互式设计至关重要。为了利用DIProT中的两个基于深度学习的模型实现这一目标,科研团队采取了两种策略。首先,使用ESMFold作为折叠预测模型。其次,使用非自回归解码范式来建立逆折叠模型,它有固定的序列生成迭代次数。相比之下,现有的自回归模型需要的迭代次数与蛋白质长度一样多。非自回归逆折叠模型的推理时间不会随着蛋白质长度的增加而有明显变化,而自回归方法的推理时间则呈线性增长。这使得DIProT比现有的自回归模型(如ProteinMPNN)快得多。
蛋白质设计是一个关键而迷人的问题,长期以来一直吸引着生物学家。在本文中,科研团队介绍了一种非自回归的逆折叠模型,该模型可用于蛋白质设计。科研团队将该模型与另一个蛋白质折叠预测模型ESMFold相结合,开发出了交互式蛋白质设计工具包DIProT。DIProT将理论知识整合到数据驱动的蛋白质设计模型中,并在序列和结构层面提供全面的分析通道,简化了用户的设计工作流程。此外,它还提供了设计结果的可视化和用户交互功能。
He J, Wu W, Wang X. DIProT: A deep learning based interactive toolkit for efficient and effective Protein design. Synth Syst Biotechnol. 2024 Feb 8;9(2):217-222. doi: 10.1016/j.synbio.2024.01.011.
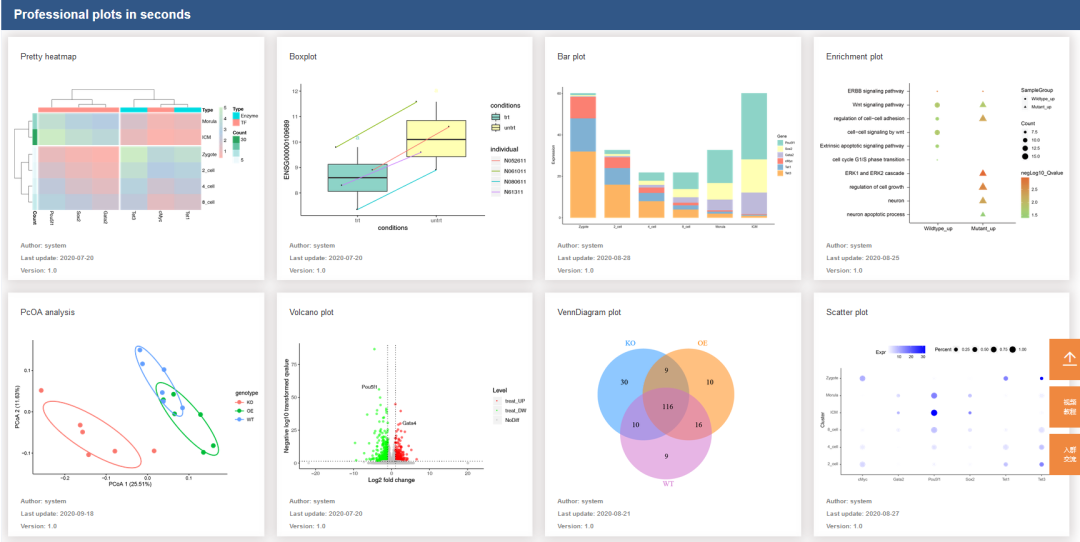
高颜值免费 SCI 在线绘图(点击图片直达)
最全植物基因组数据库IMP (点击图片直达)
往期精品(点击图片直达文字对应教程)
机器学习