在扩展长度和时间尺度上,机器学习原子间势Machine learning interatomic potentials (MLIPs) ,以接近从头计算的精度进行材料模拟。在材料的研究和设计中,机器学习原子间势发挥着广泛的应用。
近日,美国 加利福尼亚大学圣迭戈分校(University of California, SanDiego)Tsz Wai Ko & Shyue Ping Ong,在Nature Computational Science上发表评论文章,讨论了机器学习原子间势MLIPS的最新进展、挑战和前景。 Recent advances and outstanding challenges for machine learning interatomic potentials.
Recent advances and outstanding challenges for machine learning interatomic potentials.原子级模拟Atomistic simulations是研究材料的重要工具。原子模拟的可靠性,主要取决于给定势能面potential energy surface (PES) 质量。电子结构方法提供了跨周期表势能面PES的从头计算描述,但是,具有较高的计算开销和不利的系统尺寸缩放。另一方面,传统原子间势interatomic potentials (IPs),将势能面PES参数化为原子自由度函数,获得了数量级的加速和相对于原子数的线性缩放,但却牺牲了显著的精度和可转移性。
在过去二十年里,机器学习原子间势IP(MLIP)已经成为一种变革性的替代方案,在这两个极端之间架起了一座桥梁。机器学习原子间势MLIP被训练成具有其相应的能量、力和/或应力参考结构的数据集,所述能量、力和/或应力,通常从精确的从头计算获得。最先进的机器学习原子间势MLIPS,通常在能量上达到<5 MeV atom–1的预测精度,在力学上达到<0.1 eVÅ–1的预测精度,几乎比传统的原子间势IPS高出一个数量级,足以解决许多多晶型之间的较小能量差异1。
这篇评论,将讨论和比较机器学习原子间势MLIP架构的最新进展,以及突出的挑战和机遇。
机器学习原子间势MLIP,包括两个相互关联的组件:用于描述原子特征和位置特征,以及用于将这些特征映射到势能面PES的机器学习ML模型。机器学习原子间势MLIP,可大致分为两种类型(图1)。大多数机器学习原子间势MLIP,使用一组描述符来表示每个原子的局部化学环境,例如原子密度或键距和角度,以及从简单线性回归到原子核方法到神经网络的各种机器学习ML算法2。最近,出现了一类新的机器学习原子间势MLIP,利用图形描述与消息传递神经网络相结合。此后,将前者称为局域机器学习原子间势MLIPS(L-MLIPS),将后者称为图机器学习原子间势MLIPS(G-MLIPS)。
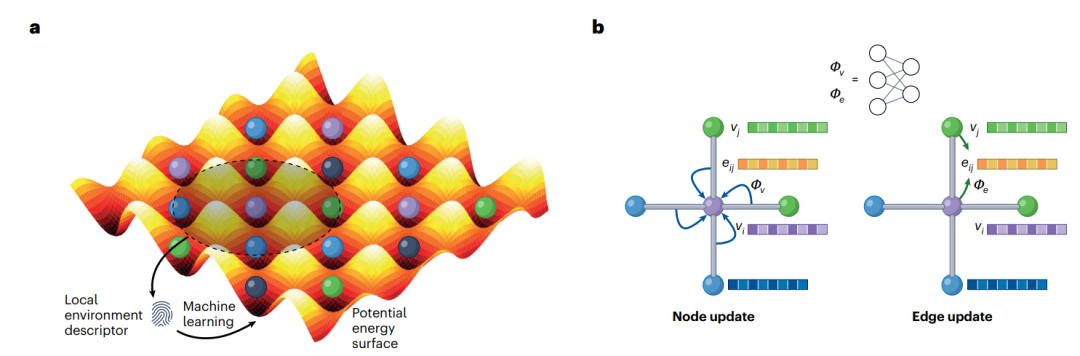
图1:机器学习原子间势的类型。
局域机器学习原子间势MLIPSL-MLIPS的主要优点是,相对紧凑性和计算效率。最近,局域机器学习原子间势L-MLIPS主要进展是原子簇扩展3,提供了一种分层的、完整的和有效的方法,以合并多体项。这有效地解决了仅具有二体和三体描述符的不同原子环境之间不完整表示4。尽管取得了重大成功,但局域机器学习原子间势L-MLIPS的关键限制是特征数量与不同元素数量的组合爆炸,这对所需训练数据量以及计算效率产生了相应的影响。因此,局域机器学习原子间势L-MLIP主要应用于包含多达四种元素的相对化学“简单”的系统。
相比之下,图机器学习原子间势G-MLIP的主要优势是,处理任意复杂系统的能力。在图机器学习原子间势G-MLIPS中,每个物种被表示为唯一的向量。因此,图机器学习原子间势G-MLIP不会遭受具有不同元素数量的相同组合爆炸5。此外,通过消息传递步骤的数量,原子相互作用的范围,可以扩展到图构造的截止半径之外。然而,在建模多体交互方面,消息传递框架效率不高。因此,大多数现代图机器学习原子间势G-MLIPS,例如材料三体图网络3-body Graph Network(M3GNet)5和消息传递原子簇扩展Message-passing Atomic Cluster Expansion (MACE)6,将消息传递图与基于传统原子间势IP或局域机器学习原子间势L-MLIPS形式的多体项相结合,以实现两全其美。此外,非局部消息传递图机器学习原子间势G-MLIP本质上比局域机器学习原子间势L-MLIP更难并行化。因此,图机器学习原子间势G-MLIP的计算强度,仍然比局域机器学习原子间势L-MLIP高一个数量级,开发能够充分利用大型高性能计算(CPU和GPU)资源的高效实现,仍然开放的挑战。
所有原子间势IP都是对称感知的。至少,在相同原子的平移、旋转和排列下,所有原子间势IP必须尊重热能面PES的不变性,这可以使用标量特征来实现。近年来,人们对将等方差的概念应用于机器学习原子间势MLIP,特别是图机器学习原子间势G-MLIP产生了极大的兴趣。等变图机器学习原子间势G-MLIP,通过输入属性和消息传递函数的设计,在坐标变换下保留了张量的变化。例如,神经等变原子间势 neural equivariant interatomic potential (Nequip),一种流行的等变图机器学习原子间势G-MLIP,使用投影到球谐基上的归一化相对位置向量作为边缘属性,并使用几何张量积,将边缘特征与相邻原子特征相结合。
等变图机器学习原子间势G-MLIPS的主要优点是,可以使用原子之间的相对位置矢量,这比不变图机器学习原子间势G-MLIPS中,使用的标量键距和角度更具表现力。在小分子系统6和一些大块系统7上,进行了一系列基准测试发现,等变图机器学习原子间势G-MLIP,可以用相当少的训练点,达到与不变图机器学习原子间势G-MLIP相当的精度。应该强调的是,这些基准测试是,在有限数量的系统上进行的,最近的研究,似乎并不支持等变机器学习原子间MLIP相对于不变机器学习原子间MLIP具有实质性的优势,特别是在大块晶体上8。公开的挑战是决定性的,最好是数学上严格的,证明在不同结构和化学系统的性能方面,等方差优于不变性的优势(或不利优势)。
大多数原子间势IP(机器学习ML或传统原子间势IP)都是针对有限元素和结构定制的。这可以归因于传统和局域机器学习原子间势L-MLIP描述符与元素数量的不利组合缩放,以及没有覆盖广泛化学范围的大型标准化数据集。随着图机器学习原子间势G-MLIPS和密度泛函DFT材料数据的大型开放数据库(如材料项目)的开发,这两个限制都得到了显著缓解。这产生了在覆盖整个元素周期表的数据集上,训练“通用” 图机器学习原子间势G-Mlips5,6,9。
在目前的发展阶段,通用和定制图机器学习原子间势MLIP服务于不同和互补的目的。虽然在给定相同的训练数据5,9情况下,图机器学习原子间势G-MLIP的性能与定制局域机器学习原子间势MLIP相当,但当前一代的通用图机器学习原子间势G-MLIP,通常在特定化学物质的准确性方面做出妥协,以换取在不同化学物质中相当好的准确性。这可能是由于现有的全周期表势能面PES数据集的相对不太严格DFT收敛标准等相关问题以及模型复杂性。事实上,最近研究表明,解决这两个问题,可以提高通用图机器学习原子间势G-MLIPS准确性和外推性10。尽管存在这些限制,在不同化学空间的材料的发现和模拟中,现有的通用图机器学习原子间势G-MLIP具有广泛的应用。并且有效地取代了速度和可扩展性优先于准确性的从头开始方法,例如对数百万种材料进行初步筛选。通用图机器学习原子间势G-MLIP也可以被认为是材料模拟的“基础模型”,即在广泛的任务中具有可靠性能的预训练模型,可以作为特定化学或应用的定制机器学习原子间势MLIP进一步快速重新训练。
无论材料体系结构如何,机器学习原子间势MLIP的准确性,都取决于所训练的数据。生成这种数据集的主要方法是,使用半局部广义梯度近似generalized gradient approximation (GGA) 泛函的密度泛函理论DFT计算。然而,众所周知,广义梯度近GGA泛函,具有可以用现代meta-GGA泛函11解决的主要缺点。使用这些修正泛函的主要障碍是,较高的计算成本。然而,使用最先进的方法生成开放、高质量势能面PES数据集是,未来机器学习原子间势MLIP开发的关键优先事项。
另一个主要优先事项是,开发具有更高数据效率的采样和训练方法。当前机器学习原子间势MLIP开发最佳实践,通常包含某种形式的主动学习active learning (AL),以确保全面覆盖感兴趣的配置空间。最近,诸多新方法(如熵最大化和不确定性驱动的动力学)已被应用于将采样偏向于未探索的空间,这加速了主动学习AL。尽管有这些进步,但对于复杂的配置空间,主动学习AL可能是非常低效的。Qi等人10最近提出了一种方法,该方法利用通用图机器学习原子间势G-MLIP,以生成较大的配置空间,而不需要密度泛函理论DFT计算,然后进行降维和分层采样,以确保全面覆盖整个配置空间。这种方法已被证明,可以减少并在某些情况下消除对迭代主动学习AL的需要。最后,迁移学习和多保真方法,还可以显著减少所需的高保真训练数据点的数量12。
除了几个值得注意的例外9,13,14,大多数机器学习原子间MLIP架构,没有明确地包含原子电荷和磁性。对于广泛的材料科学问题,准确描述这些长程相互作用,是必要的,包括反应系统和界面、电子驱动相变、磁性材料等。将电荷和磁性结合到机器学习原子间势MLIPS中的主要挑战,不在于架构开发,而在于训练数据的生成和计算效率。几种有效的方法,如电荷平衡方案和自旋-晶格图机器学习原子间势G-MLIP,已经被开发出来。然而,除了耦合的结构和化学自由度之外,训练这样的机器学习原子间势MLIP,需要包括电荷和磁性自由度的数据集,这极大地增加了计算成本和数据生成的复杂性。此外,结合静电和磁相互作用所增加的复杂性,也往往会对机器学习原子间势MLIP的计算效率和缩放产生不利影响,从而减少可以应用的长度和时间尺度13。实际的算法改进是必要的,以优化这种模型和一般机器学习原子间势MLIP的效率,以扩大其适用范围。
总之,机器学习原子间势MLIPS,通过提供一种强大的、自动化的方法,从而参数化势能面PES,对材料科学产生了变革性的影响。在超出从头计算方法可达到的长度和时间尺度上,,机器学习原子间势MLIPS模拟材料,同时再现接近从头计算的精度。最近,通用图机器学习原子间势G-MLIP的出现,进一步扩展了其应用范围,从选择化学研究扩展到广泛的材料发现。在功能、架构和最佳实践方面的持续进步,将使机器学习原子间势MLIP具有更高的准确性、效率和可推广性。然而,这是机器学习原子间势MLIP软件实现的优化。特别是图机器学习原子间势G-MLIP,在计算上仍然比传统的原子间势IP昂贵几个数量级,这可能会限制应用的长度和时间尺度。
未来机器学习原子间势MLIP发展的主要瓶颈,最终是势能面PES数据的稀缺。生成高质量的势能面PES数据,特别是使用最先进的函数,是创建机器学习原子间势MLIP最昂贵的步骤。现有材料数据库的开发,主要侧重于材料发现和设计的属性信息;对生态系统服务付费综合数据的开发、管理和传播的关注相对较少。因此,开发可查找、可访问、可互操作和可重复使用FAIR的势能面PES数据存储库15,作为对OpenKim等原子间势IP数据库补充,是关键优先事项。势能面PES数据库,不仅应包括典型的势能面PES信息,还应包括辅助信息(如电荷密度、磁矩等),这些辅助信息来源于未来模型开发中的电子结构计算。可以作为开发机器学习原子间势MLIP通用基准的基础,为训练、测试和评估不同机器学习原子间势MLIP架构的计算效率,提供标准化数据集。
1. Zuo, Y. et al. J. Phys. Chem. A 124, 731–745 (2020).2. Behler, J. & Csányi, G. Eur. Phys. J. B 94, 142 (2021).3. Drautz, R. Phys. Rev. B 99, 014104 (2019).4. Musil, F. et al. Chem. Rev. 121, 9759–9815 (2021).5. Chen, C. & Ong, S. P. A. Nat. Comput. Sci. 2, 718–728 (2022).6. Batatia, I., Kovacs, D. P., Simm, G., Ortner, C. & Csányi, G. Adv. Neural Inf. Process. Syst. 35, 11423–11436 (2022).7. Batzner, S. et al. Nat. Commun. 13, 2453 (2022).8. Riebesell, J. et al. Preprint at https://arxiv.org/pdf/2308.14920.pdf (2023).9. Deng, B. et al. Nat. Mach. Intell. 5, 1031–1041 (2023).10. Qi, J. et al. Preprint at https://arxiv.org/abs/2307.13710 (2023).11. Sun, J. et al. Nat. Chem. 8, 831–836 (2016).12. Smith, J. S. et al. Nat. Commun. 10, 2903 (2019).13. Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A. Nat. Commun. 12, 398 (2021).14. Yu, H. et at. Preprint at https://arxiv.org/abs/2203.02853 (2023).15. Wilkinson, M. D. et al. Sci. Data 3, 160018 (2016)Ko, T.W., Ong, S.P. Recent advances and outstanding challenges for machine learning interatomic potentials. Nat Comput Sci (2023). https://doi.org/10.1038/s43588-023-00561-9https://www.nature.com/articles/s43588-023-00561-9声明:仅代表译者个人观点,如有不科学之处,请在下方留言指正!









