尽管计算能力有所提高,但元素和晶体结构类型组合的巨大空间,使得稳定材料的大规模高通量,依然非常昂贵,特别是对于复杂材料和受环境条件(如有限温度)影响的材料。
当基于物理计算方法和劳动密集型实验不可行时,机器学习 machine learning (ML) 方法,可成为一种快速而强大的替代方法。由于丰富的实验和第一性原理数据,以及为材料建模设计的修正机器学习ML框架,在预测稳定性参数和加速发现新的稳定材料方面,机器学习ML证明是有效的。近日,美国 西北大学(Northwestern University)Sean D. Griesemer, Yi Xia & Chris Wolverton,在Nature Computational Science上发表综述文章,总结了应用最大似然方法预测材料稳定性的最新进展,特别是零温和有限温度稳定性的预测。还强调了在预测其他热力学参数(如压力和表面/界面能)时,需要进行更多的机器学习ML开发,这实际上会影响材料的稳定性。 Accelerating the prediction of stable materials with machine learning.
Accelerating the prediction of stable materials with machine learning. 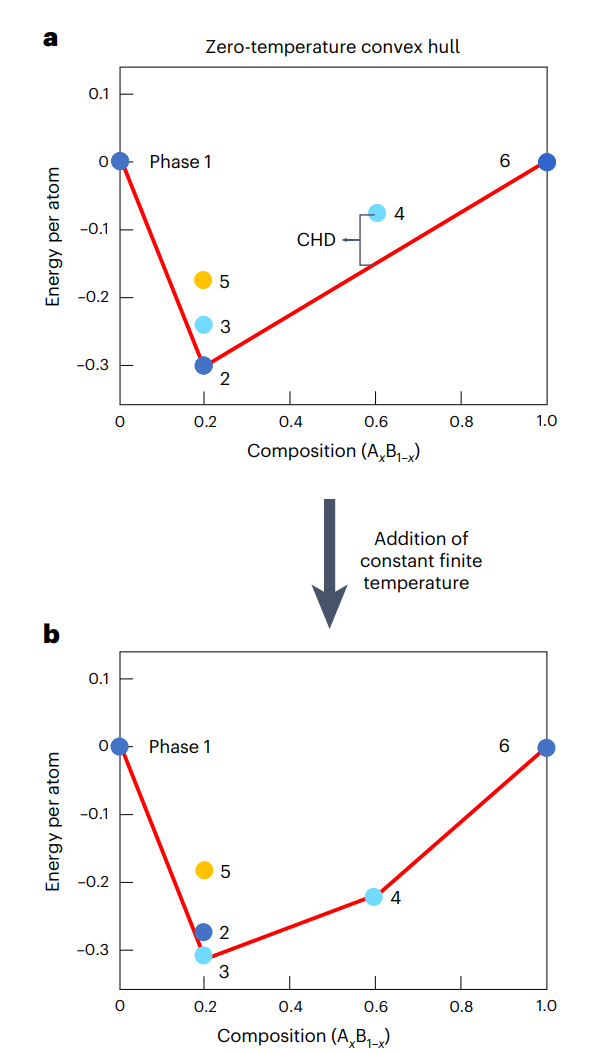
图1: 随温度变化的计算几何凸包convex hull示意图。
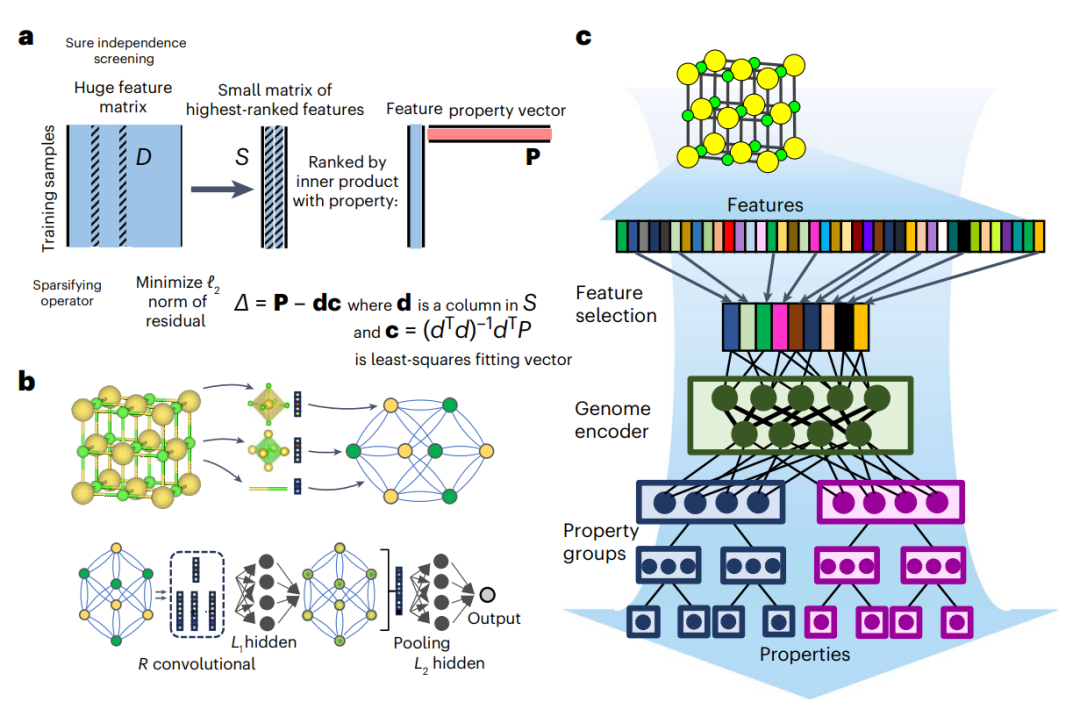
图2: 材料稳定性预测的最新机器学习machine learning,ML框架示例。
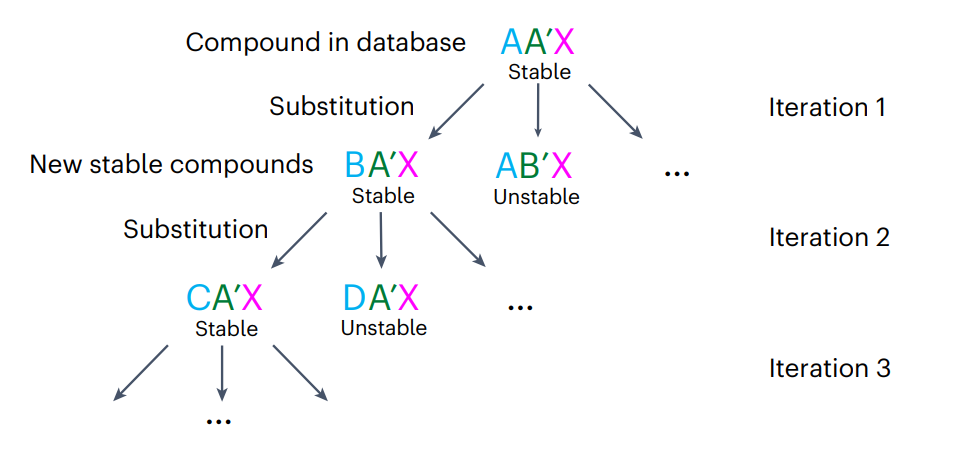
图3: 基于元素取代法,新稳定化合物研发的工作流程。
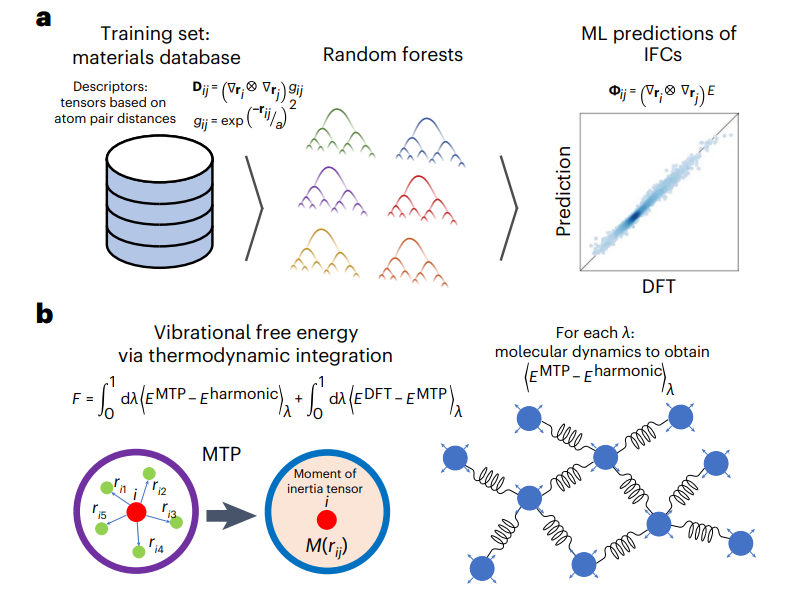
图4: 在T>0时,最新机器学习ML模型示例,用于预测有序化合物的振动自由能。
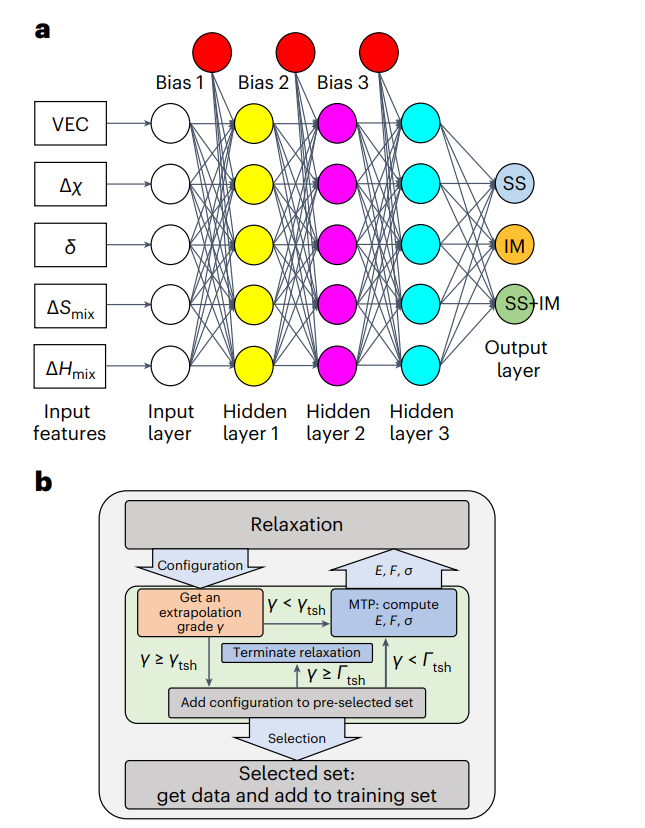
图5 :在T>0时,预测无序化合物的最新机器学习ML模型的例子。
Griesemer, S.D., Xia, Y. & Wolverton, C. Accelerating the prediction of stable materials with machine learning. Nat Comput Sci (2023). https://doi.org/10.1038/s43588-023-00536-whttps://www.nature.com/articles/s43588-023-00536-w声明:仅代表译者个人观点,小编水平有限,如有不当之处,请在下方留言指正!
近日,美国 劳伦斯伯克利国家实验室(Lawrence Berkeley National Laboratory)Xingyi Guan, Teresa Head-Gordon等,在Nature Computational Science上发文,报道训练了等变机器学习equivariant machine learning (ML)模型,用以预测有限温度和压力条件下,氢气燃烧的能量和力。
反应化学的这极具有挑战案例表明,由于过度依赖对训练重要数据的化学直觉,机器学习ML势能面很难完成。相反,使用元动力学metadynamics,作为主动学习工作流程的一部分的“负设计negative design”数据采集策略,有助于创建避免不可预见高能或非物理能量配置的机器学习ML模型。该项策略,更快地收敛势能面。在时间尺度上,当委员会查询query-by-committee models模型与进一步的分子动力学不一致时,现在可以更有效地调用外部从头算资源,而无需进行机器学习ML再训练。通过混合机器学习ML-物理模型,实现了成本降低两个数量级,预测几个氢燃烧反应过渡态机制中的自由能变化。 Using machine learning to go beyond potential energy surface benchmarking for chemical reactivity.
Using machine learning to go beyond potential energy surface benchmarking for chemical reactivity. 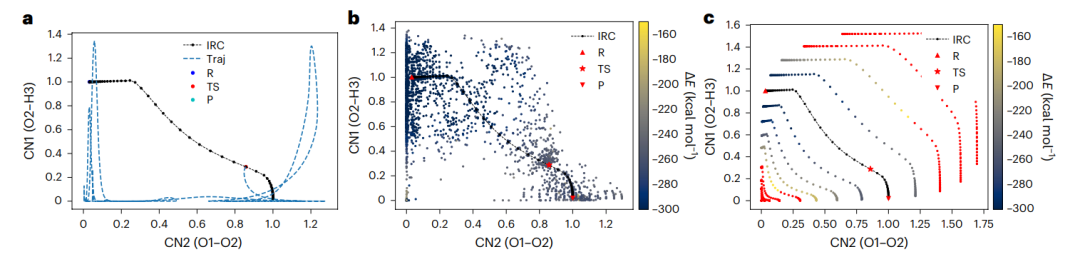
图1:在氢燃烧的机器学习machine learningML模型中,缺失数据的观察和膨胀数据的添加。
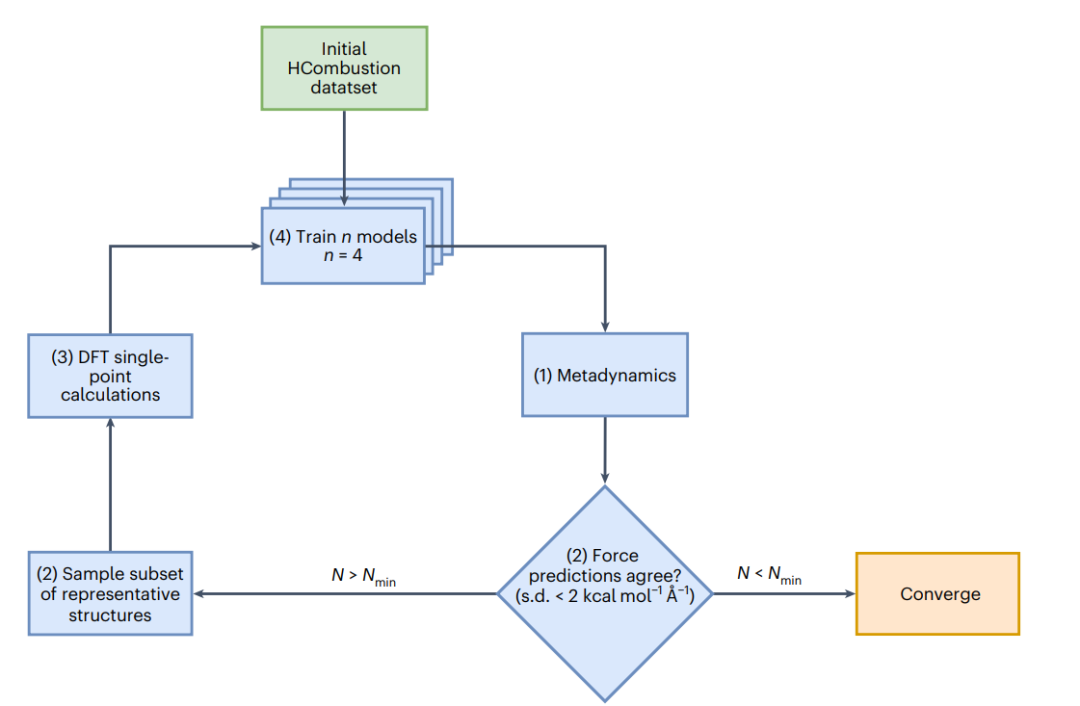
图2:基于委员会查询query-by-committee和元动力学模型,主动学习active learning,AL工作流程示意图。
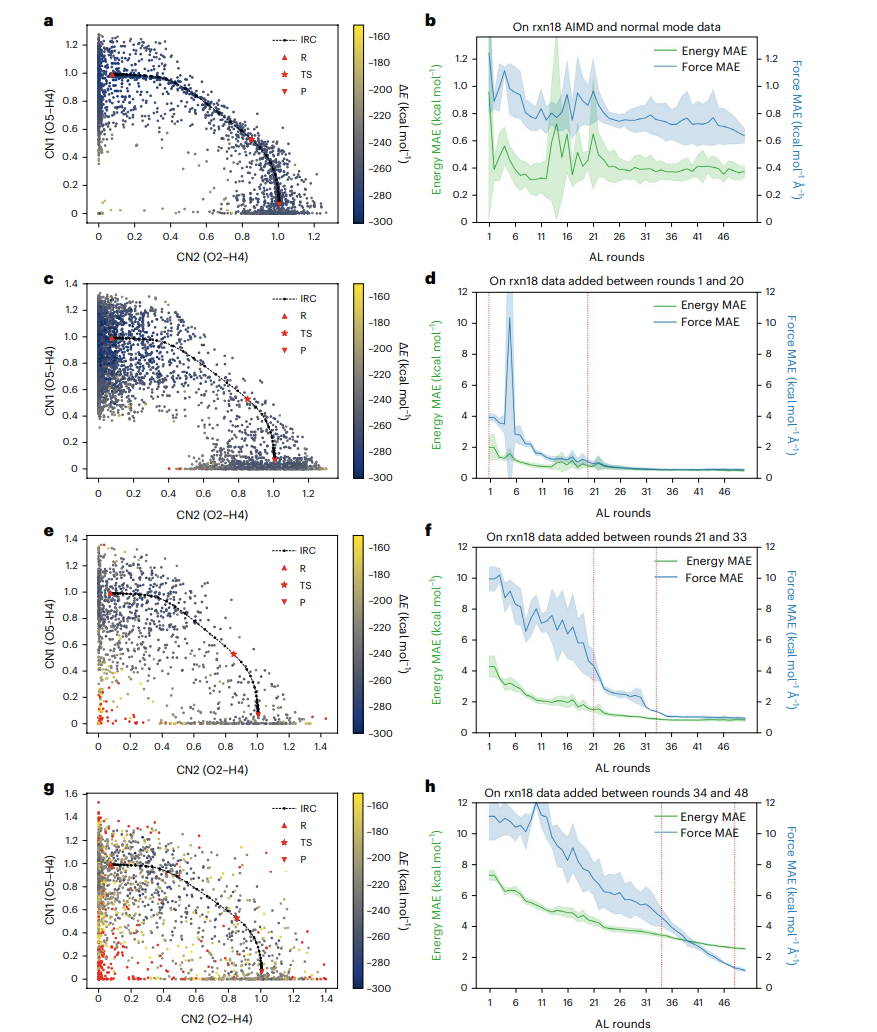
图3: rxn18算法的主动学习AL数据和错误。
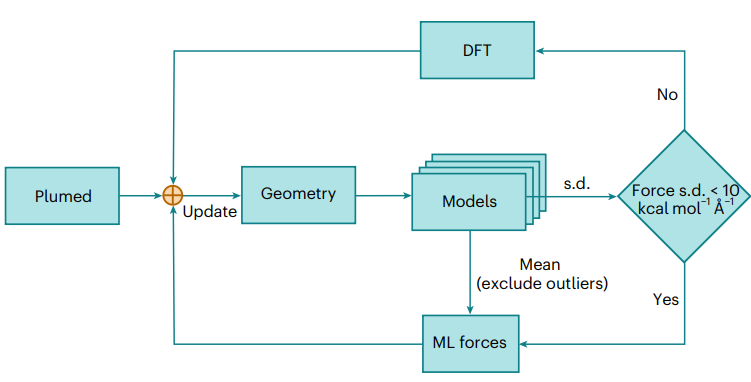
图4: 自由能面重建的新工作流程示意图。
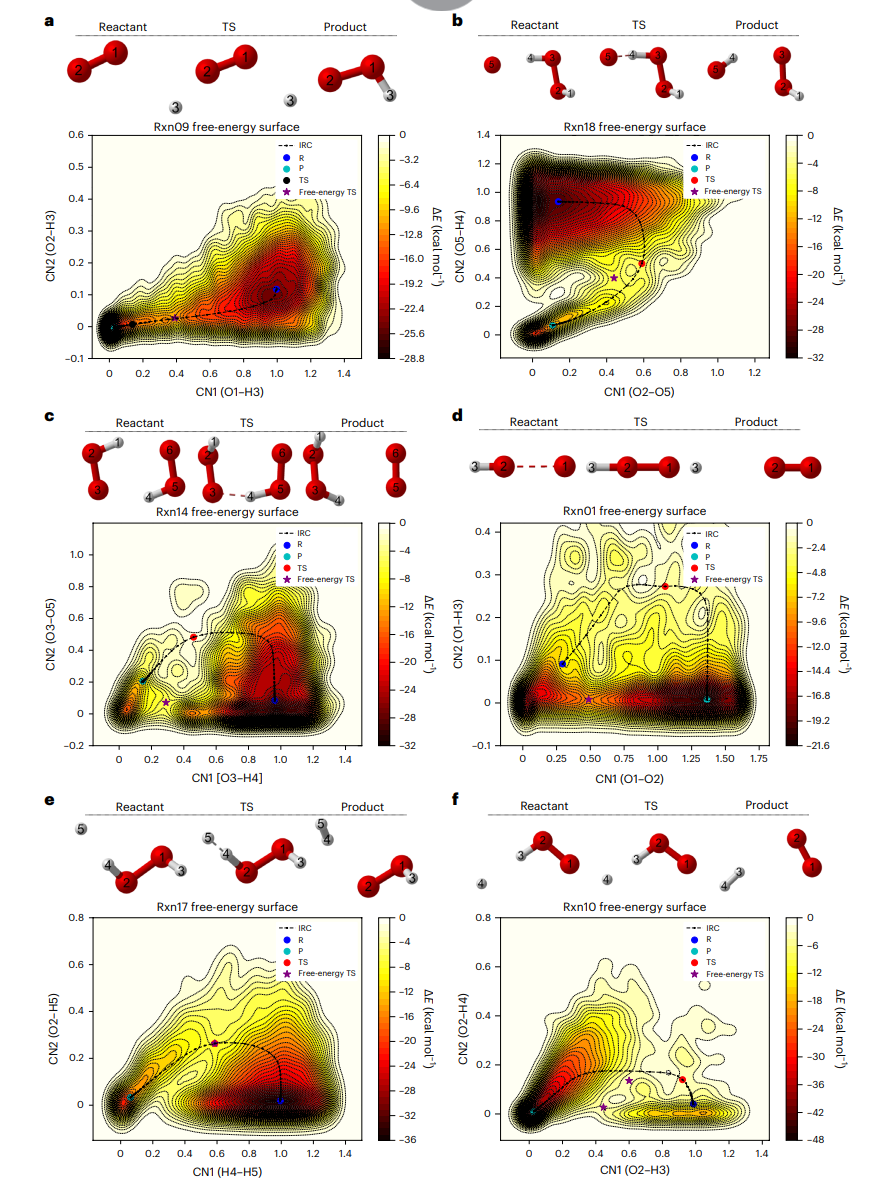
图5: 基于氢燃烧的混合模型,从元动力学重建的自由能面。
Guan, X., Heindel, J.P., Ko, T. et al. Using machine learning to go beyond potential energy surface benchmarking for chemical reactivity. Nat Comput Sci (2023). https://doi.org/10.1038/s43588-023-00549-5https://www.nature.com/articles/s43588-023-00549-5声明:仅代表译者个人观点,小编水平有限,如有不当之处,请在下方留言指正!







