将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

编辑 | 紫罗
用有机溶剂溶解聚合物是高分子材料研究和开发中必不可少的过程,包括塑料回收、聚合物合成、精制、涂漆和涂层等。然而,预测和理解聚合物-溶剂二元系统的相平衡或相分离是聚合物化学中尚未解决的基本问题。
日本统计数理研究所(The Institute of Statistical Mathematics)的研究人员利用三菱化学集团 (MCG) 的量子化学计算数据库,开发了一种新型机器学习系统,用于确定任何给定聚合物与其候选溶剂的混溶性,称为 χ 参数。
该系统使科学家能够通过使用高通量量子化学计算整合计算机实验产生的大量数据,克服聚合物与溶剂混溶性实验数据有限所产生的限制。
该研究以「Multitask Machine Learning to Predict Polymer–Solvent Miscibility Using Flory–Huggins Interaction Parameters」为题,发表在《Macromolecules》杂志上。
论文链接:https://pubs.acs.org/doi/10.1021/acs.macromol.2c02600
需要一个 χ 参数的数据集来训练模型
预测和理解聚合物溶剂溶液中的相平衡或相分离代表了聚合物科学中尚未解决的基本问题。聚合物混溶性的相行为和热力学取决于与溶剂混合的具有一定分子量分布的聚合物的分子间和分子内相互作用。
根据 Flory-Huggins 聚合物溶液理论,聚合物溶液的热力学性质,例如混溶性或溶胀平衡,可以用称为 Flory-Huggins χ 参数的聚合物-溶剂相互作用参数来表示。对于给定的 χ 参数值,聚合物-溶剂相空间可以描述为以下可控变量的函数:温度、体积分数和分子链长度。然而,通过实验测量 χ 参数在技术上困难且成本高昂。
尽管已经开发了各种模型来计算预测 χ 参数,但基于聚合物和溶剂溶解度参数之间的距离的经验模型是最广泛使用的。例如,汉森溶解度参数 (Hansen solubility parameter,HSP) 将给定分子的潜在溶解度表示为由色散(范德华力)、极性(偶极矩)和氢键成分组成的三维矢量。聚合物-溶剂溶解度是根据 HSP 向量之间的距离确定的。基于量子化学的 COSMO-RS 方法已应用于广泛的聚合物-溶剂体系。然而,这些原子模拟的计算成本很高。
近年来,随着大数据和高性能计算资源的可用性不断增加,机器学习已成为实现高速预测的一种有前途的方法。然而,它需要一个 χ 参数的数据集来训练模型。在这方面,已经提出了多种技术来获得实验 χ 参数值。然而,此类技术在技术上困难且成本高。此外,每种方法的适用性都有限。这使得创建可应用于各种系统的高度通用的 χ 参数预测模型变得困难。
目前,利用机器学习研究聚合物混溶性方面进展甚微。尽管已有一些研究,但已有模型的适用范围是有限的,因为训练数据在数量上是有限的。
机器学习框架,实现高度通用和稳健的 χ 参数预测
在此,研究人员提出了一个机器学习框架,以实现高度通用的温度相关 χ 参数预测。
该模型将 χ 参数描述为聚合物和溶剂化学结构的函数,该模型使用了 1190 个实验观察到的 χ 参数样本,其中有 766 个独特的聚合物-溶剂对,由 46 个聚合物和 140 个溶剂组成。
与之前的研究一样,该数据集的化学多样性有限;此外,实验 χ 参数存在偏差。
为了克服这些限制,创建了两个辅助数据集。研究人员从 PoLyInfo 中提取了一个辅助数据集,以提供 29777 个可溶性和不溶性聚合物-溶剂对的列表。此外,使用 COSMO-RS 进行量子化学计算,生成了 9575 个聚合物-溶剂对的 χ 参数内部数据集。
研究人员使用这三个数据集,进行了基于深度神经网络的多任务机器学习,以同时进行聚合物混溶性的二元分类以及真实系统和模型系统中 χ 参数的定量预测。
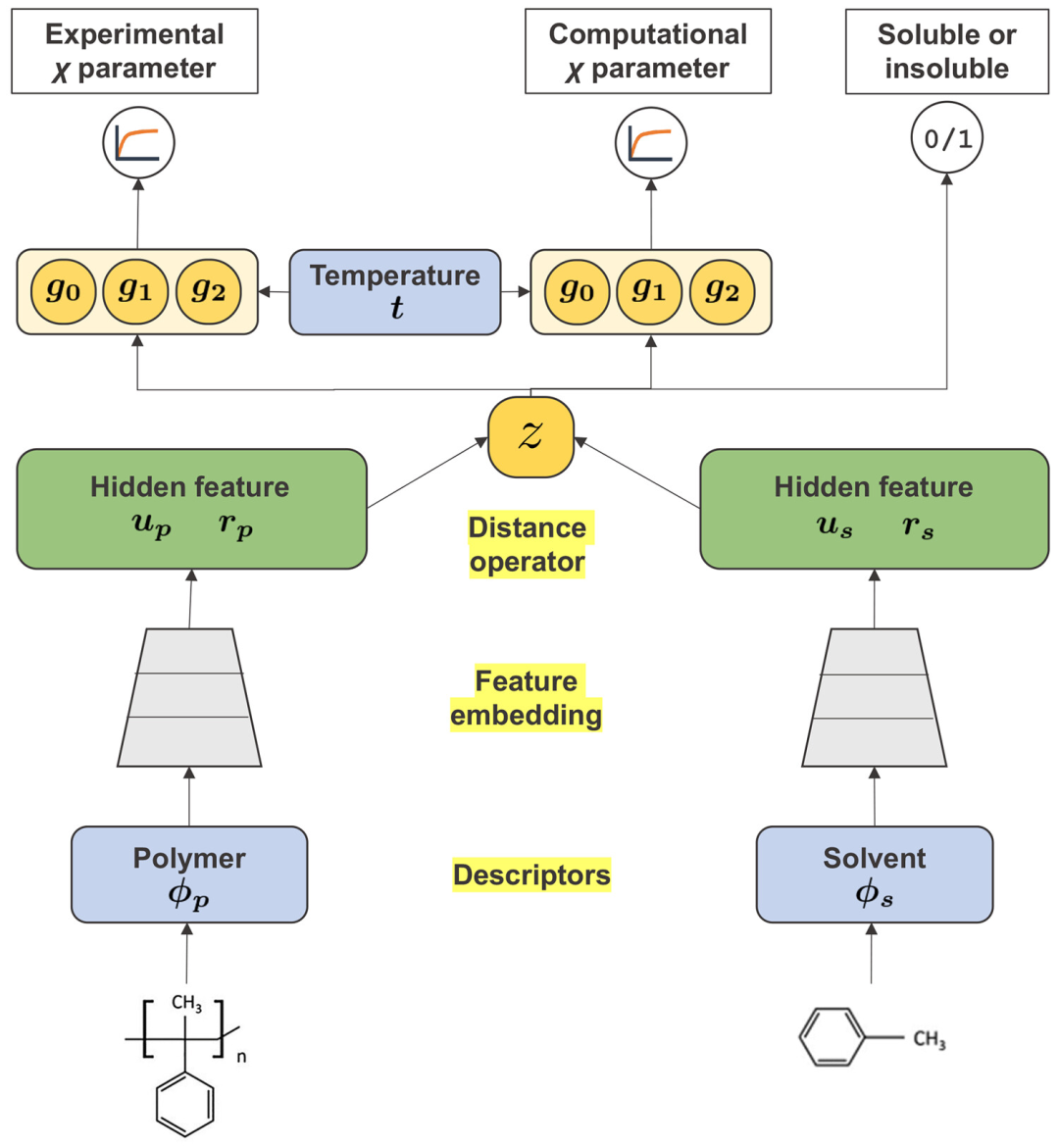
图示:用于预测暴露于溶剂 s 的聚合物 p 的 Flory-Huggins χ 参数的神经网络架构。(来源:论文)
神经网络的输入变量包括聚合物-溶剂对及其化学结构以及混合温度。输出层分为三个不同的任务:实验和计算的 χ 参数的预测值以及指示给定聚合物-溶剂对是否可混溶的分类概率。从输入到输出的映射是使用多层神经网络建模的,保留了与汉森溶解度球(Hansen’s solubility sphere)的类比。虽然 HSP 距离是在与色散、极性和氢键相关的三种不同力的空间中定义的,但所提模型旨在通过将聚合物-溶剂化学特征嵌入到 10-40 维的潜在空间中,自主创建广义的、扩展的溶解度球。嵌入的特征及其相关维度是根据观察到的数据自主学习的。
训练后的模型表现出相当好的泛化性能。预测能力超过了使用 COSMO-RS 的预测和使用汉森溶解度球的预测器。由于聚合物溶剂种类的结构多样性不足,实验 χ 参数的数据集提供的训练样本有限。因此,在普通的单任务机器学习范围内,训练模型的适用范围仅限于狭窄数据分布的内部或稍外部。这里表明,通过与另外两个大数据集联合学习,可以成功扩展模型的适用范围。
计算速度比传统量子化学计算快约 40 倍
在这项研究中,研究人员创建了一个基本模型,可以同时解决聚合物混溶性的三个密切相关的任务。
研究人员开发的预测模型计算 χ 参数的速度大约是传统量子化学计算的 40 倍。使用该模型,可以超高速地筛选数百万数量级的候选溶剂分子。
在计算时间方面,在传统服务器上执行量子化学计算并为 47 种聚合物和 138 种溶剂创建 COSMO 文件总共花费了 4129 秒。此外,从 COSMO 文件中计算 1190 对的 χ 参数需要 732 秒。因此,每个聚合物-溶剂对需要 (4129 + 732)/1190 ≃ 4.1 s。这比神经网络的执行时间慢了近 40 倍,神经网络的执行时间约为每个聚合物-溶剂对 0.11 秒,包括描述符计算。
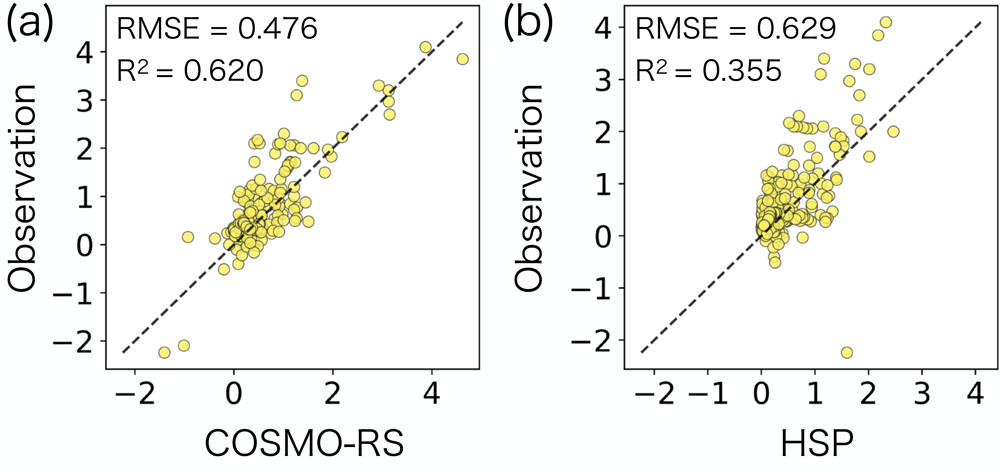
图示:通过 (a) 使用 COSMO-RS 方法进行量子化学计算和 (b) HSP 距离预测器来预测实验 χ 参数。(来源:论文)到目前为止,该模型已被证明是准确的,当它涉及到需要什么来使聚合物和溶剂成为一种适合回收的均匀混合物时,需要大量的猜测工作和试验和错误,以创造一种适合回收方法的混溶物质。
但对于任何新兴技术,在真正准备好大规模使用之前,总是可以做一些工作来简化流程并解决问题。
该研究共同作者 Ryo Yoshida 说:「为了进一步改进和扩展机器学习技术,促进材料信息学领域的开放创新和开放科学,我们已将开发的部分源代码和数据向公众开放。」
为了便于后续的研究,Python 源代码和其他相关资料已经上传到 GitHub 上。预计这些结果将有助于克服聚合物科学领域中尚未解决的重要问题。
GitHub 地址:https://github.com/yoshida-lab/MTL_ChiParameter
参考内容:https://phys.org/news/2023-10-machine-reveals-dissolve-polymeric-materials.html
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。









