将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


优化分子(材料或药物)的特性以获得更强的韧性、更低的毒性或更好的生物利用度一直是一个长期存在的挑战。
华为与复旦大学的联合研究团队提出了一种名为 Q-Drug(药物的量子启发优化算法)的分子优化框架,该框架利用量子启发算法来优化离散二元域变量上的分子。
该框架首先使用离散 VAE 将分子编码为二进制嵌入。然后使用二进制嵌入来构造类似伊辛能量的目标函数,并采用最先进的量子启发优化算法来寻找最优值。解码对应于最优值的二进制嵌入以获得优化的分子。
研究人员测试了优化药物分子特性的框架,发现它优于其他分子优化方法,找到具有更好特性的分子所需时间是之前所需时间的 1/20 至 1/10。
并且,该框架还可以直接部署在各种量子计算设备上,例如激光脉冲CIM、FPGA Ising Machine和基于量子退火的量子计算机等。
该研究以「Q-Drug: a Framework to bring Drug Design into Quantum Space using Deep Learning」为题,于 2023 年 8 月 25 日发布在 arXiv 预印平台。

研究背景
当化学分子(例如药物或材料)需要改善性能(例如更低的毒性、更好的生物利用度和更高的韧性)时,分子优化是必要的。修改分子的结构可以实现这些结果,但这是一个费力且成本高昂的过程,需要进行多次试错尝试。人们已经开发了许多机器学习模型来优化分子,包括条件 VAE、JT-VAE 和 GCPN,它们分别是基于条件生成、贝叶斯优化和强化学习等不同优化算法的代表性模型。
尽管这些方法已经取得了一些进展,但它们仍然面临许多挑战,例如花费太多时间并陷入局部极小值,这会对优化结果产生负面影响。此外,以前的分子优化方法侧重于连续域的优化,这可能无法适当地解决分子活性悬崖等问题。因为当活性悬崖发生时,分子结构的细微变化可能会导致效力的巨大差异。研究人员的目标是将分子优化方法扩展到离散领域。
为了探索基于量子的离散优化算法用于分子优化的可能性,研究人员可以将分子编码为二进制嵌入,对这些嵌入进行优化,然后将它们解码回分子。变分自动编码器 (VAE) 是实现这一目标的理想选择。
VAE 是一类深度生成模型,可以对数据样本进行编码并根据新代码生成(解码)样本。它们由两项组成,称为重建项和正则化项。重建项确保编码准确性,而正则化项确保从分布中采样可以生成(解码)有效的数据样本。重新参数化技巧使 VAE 变得流行。该技术确保对特定分布进行采样,并且神经网络能够正确反向传播。
目前该方法有两个主要挑战。首先,具有离散潜在变量的 VAE 很难有效地训练,因为通过离散变量进行反向传播通常是不可能的。应对这一挑战的主要方法的基本思想是找到一个定义在连续域上的分布,该分布可以逼近所需的离散分布。其次,从阶乘伯努利分布中采样以 0.5 概率取 0 或 1 的值的噪声太大,无法重建有效的数据样本。因此,研究人员引入受限玻尔兹曼机(RBM)作为生成模型,用于学习编码器输出的分布并减少采样代码中的噪声。
通过使用连续松弛和受限玻尔兹曼机 (RBM) 进行的改进,分子现在可以有效地编码为二进制形式的向量(二进制嵌入)。从伯努利分布中采样可以解码有效的分子。通过收集一批分子的属性,研究人员可以通过这些分子的二元嵌入来构建该属性的预测矩阵分解模型。矩阵分解自然采用伊辛问题的形式,可以通过量子退火有效解决。
量子启发优化算法
在这项研究中,华为团队使用了一种名为模拟分叉(SB)的量子启发优化算法(Q-Drug)来加速这个组合优化问题。SB 的起源是伊辛问题,其中涉及寻找使能量最小化的自旋构型(伊辛哈密顿量)。使用 SB 算法解决 Ising 问题会得到最优值和最优自旋配置(二进制嵌入)。解码二进制嵌入可以获得具有相应属性的最佳分子。
以此为基础,该团队构建了一个将分子编码为二元嵌入的框架,可用于构建类似 Ising 的函数来拟合分子的属性。该函数可以进一步用作属性优化问题的目标函数。与使用传统分子优化方法相比,通过在量子计算机或传统计算机上的量子启发算法中使用量子退火,研究人员可以找到更好的全局最优值并使用更少的时间。
总体而言,如图 1 所示,该方法涉及在名为 ZINC-250k 的大量药物分子数据集上对离散 VAE 进行预训练。这一步骤有助于将分子编码为二进制代码,然后解码为有效的药物分子。使用量子启发的优化算法,研究人员操纵二进制代码来最小化或最大化目标函数,从而优化二进制域中的分子。
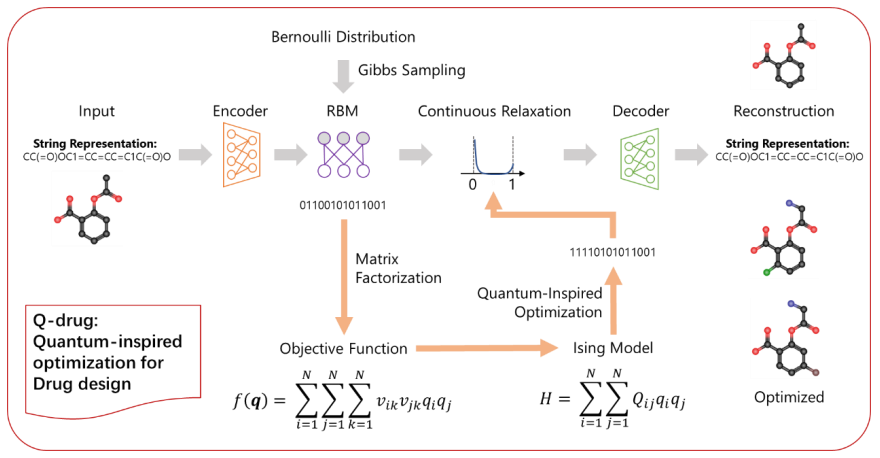
图1:量子启发分子优化(Q-drug)的框架。(来源:论文)离散VAE可以有效地编码和解码分子
华为团队想要为分子设计一个好的编码方案,它可以将分子转换为二进制嵌入,并从随机二进制嵌入中重建有效的分子。为了建立基线,研究人员尝试使用广泛使用的 VAE(变分自动编码器)将分子编码为连续嵌入。研究人员使用的数据集是 ZINC-250k,其中包括大约 250, 000 个药物分子。
VAE 的一个简单直观的扩展是 Bernoulli VAE,它直接将先验潜在空间的高斯分布更改为 Bernoulli 分布。离散 VAE 是一种更复杂的模型,它使用连续松弛和 RBM(受限玻尔兹曼机)生成。
表1:不同编码方案的比较。(来源:论文)
Q-Drug 在不同任务上均优于之前的分子优化模型
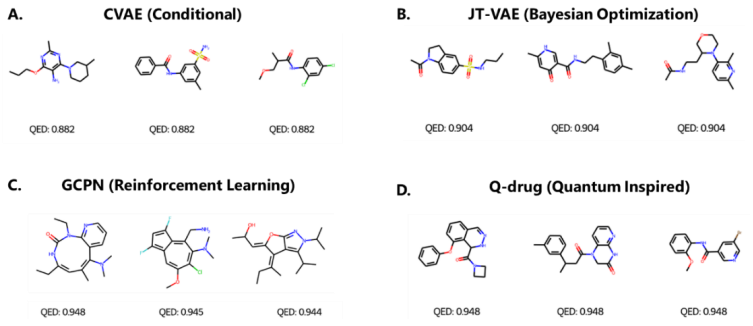
图2:使用四种不同方法在药物相似性 (QED) 优化中生成的分子样本。(来源:论文)
该团队比较了分子优化的三种优化模型:条件 VAE、JT-VAE 和 GCPN,它们分别使用条件生成、贝叶斯优化和强化学习。在 QED 药物相似性优化任务上,Q-drug 和 GCPN 可以达到 0.948 的 QED 值,这是 QED 值的实际上限,尽管它的范围在 0 到 1 之间。
然而,如果检查图 2C 中的分子,会发现 GCPN 可能只是利用了 QED 的缺陷来获得高 QED,因为该分子显然对于具有药物化学背景的研究人员来说并不理想。相比之下,CVAE、JT-VAE 和 Q-drug 生成的结构更加合理,其中 Q-drug 的优化效果最好。在比较这些模型的推理时间成本时,可以发现条件 VAE 速度很快,但优化效果较差。另一方面,Q-drug只需要贝叶斯优化和强化学习所需时间的约1/20到1/10即可达到最佳优化效果。
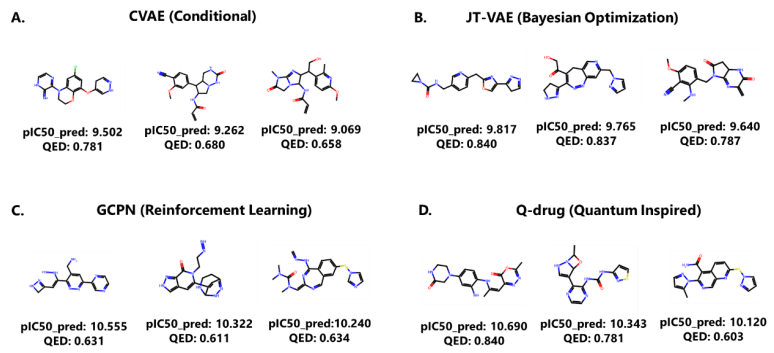
图3:使用四种不同方法在 FGFR3 生物活性和 QED 优化中生成的分子样本。(来源:论文)
对于同时优化生物活性和 QED 药物相似性的多目标优化任务,研究人员通过将 10 倍 QED 值添加到 pIC50 值,将这两个指标缩放到相同范围。结果表明,采用量子启发优化方法的 Q-drug 在两个药物靶点:FGFR3 和 FGFR4 任务上均优于其他方法。
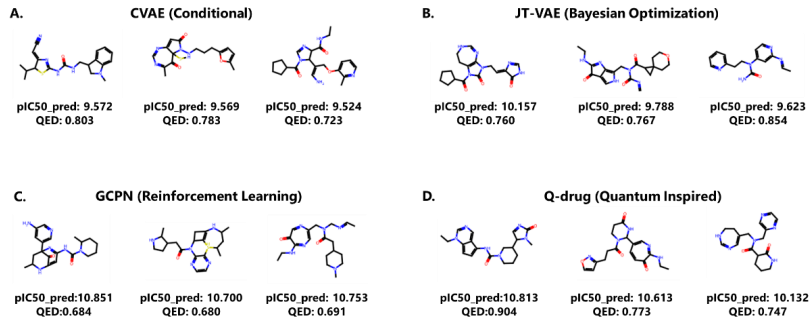
图4:使用四种不同模型在 FGFR4 生物活性和 QED 优化中生成的分子样本。(来源:论文)结语
我们周围的世界是量化的或离散的,这意味着它具有独特的、不可分割的单位。然而,传统的分子优化方法侧重于在连续域上进行优化,这可能无法适当解决分子中的“活性悬崖”等问题。华为团队的研究探索了在离散域上优化分子的可行性和前景。
研究人员展示了使用离散 VAE 编码分子的概念证明,但也承认使用 DVAE 等技术进一步改进的潜力。为了解决构建的类伊辛能量目标函数,除了量子启发的优化算法之外,研究人员建议利用基于量子计算的设备。这包括超导量子计算机、基于激光脉冲的 CIM(相干伊辛机)和基于 FPGA(现场可编程门阵列)的伊辛机。
使用 HOFM(高阶分解机)或 BOX-QUBO(使用交叉熵方法和 QUBO 的黑盒优化)构建目标函数也显示出潜力,并且适合通过量子启发算法进行优化。该研究提供了一个名为 Q-Drug 的实用框架,它利用人工智能将药物设计带入量子空间,为基于量子计算概念的更好的分子设计技术提供了新的可能性。此外,分子构象生成和分子对接等其他应用也可能从量子启发算法的优化中受益匪浅。
论文链接:https://arxiv.org/abs/2308.13171
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关
注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。









