将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

探究核空间中基因组的组织是生物学的一个重要前沿研究。Hi-C 和 Micro-C 等染色体构象捕获方法可生成全基因组染色质接触图,提供包含有关基因组结构的定量和定性信息的丰富数据。大多数全基因组染色体构象捕获数据的传统方法仅限于分析预定义的特征,因此可能会错过重要的生物信息。一个限制是,生物学上的重要特征可能会被数据中的高水平技术噪音所掩盖。
伦敦帝国理工学院(Imperial College London)的研究人员介绍一种基于复制的染色质构象接触图深度学习方法。使用暹罗网络配置,该方法可以区分技术噪声和生物变异,并且在一系列生物系统中优于图像相似性度量。在粘连蛋白和 CTCF 扰动后从 Hi-C 图谱中提取的特征分别反映了粘连蛋白和 CTCF 在结构域和边界形成中的不同生物学功能。
该研究以「A deep learning method for replicate-based analysis of chromosome conformation contacts using Siamese neural networks」为题,于 2023 年 8 月 17 日发布在《Nature Communications》。

真核染色质在细胞核内进行空间组织,以促进基本的基因组功能,包括转录、复制、修复和染色体分离。影响核结构的突变可能导致疾病。该组织的各个方面,例如染色体区域的形成、异染色质与常染色质的分离以及拓扑关联域 (TAD) 的形成,都是高度保守的,但细节更精细,例如染色质环和接触域的强度可以以细胞类型特异性的方式变化。虽然控制染色质组织的精确机制仍然是一个深入的研究领域,但形成结构域和环的主要机制之一是粘连蛋白复合物主动挤出染色质,这受到 CTCF 结合的限制。
用于绘制基因组组织图谱的关键工具包括 Hi-C 和 Micro-C。两者都结合了基于邻近的连接和高通量测序来生成全基因组染色质接触图,提供丰富的数据源,其中包含有关基因组结构的定量和定性信息。Hi-C 和 Micro-C 数据的分析通常依赖于目视检查以及对染色质构象图的已知特征进行评分的方法,例如绝缘分数、方向性指数、TAD、环或条纹。这种方法不仅费力,而且还可能导致忽略具有生物学重要性的特征。需要新的方法来分析全基因组染色质构象捕获数据中包含的大量信息。
一种非常适合大量数据的方法是使用深度学习来查找染色质构象数据的差异。于是,问题变成了识别很大程度上异质的基因组之间的细微差异,并解释构象接触图中存在的非常高水平的不均匀噪声。将这些构象图视为图像可以使用图像分析技术。Hi-C 和 Micro-C 数据包含标准图像数据集中不存在的高水平噪声,目前正在讨论用于分析此类数据的朴素图像相似性度量的适用性。
为了解决这个问题,伦敦帝国理工学院(Imperial College London)的研究人员开发并验证了 Twins,这是一种基于深度学习的分析方法,利用连体卷积神经网络进行重复。与标准图像分析工具不同,Twins 使用对比学习来区分技术噪声(重复之间的差异)和生物变异(条件之间的差异)。
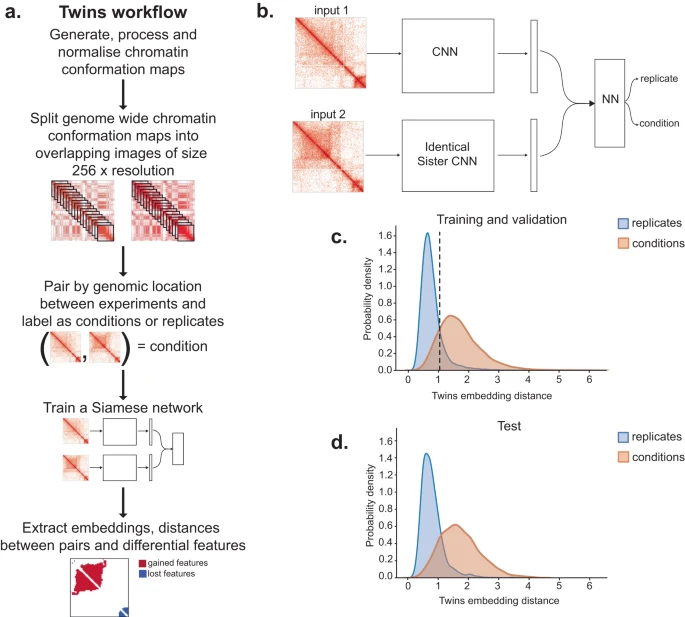
图示:一种基于复制的染色质构象接触图深度学习方法。(来源:论文)使用这种方法,该团队证明使用基于重复的机器学习,可以在染色质构象捕获数据上产生信息丰富的结果。基于复制的训练可以在多种生物环境中产生有意义的嵌入距离,包括识别 T 细胞发育过程中的细微差异。
此外,研究人员发现使用重复足以防止由于强制训练而可能出现的错误或夸大差异。与朴素的图像相似性度量(其中一些过去已应用于 Hi-C 分析)相比,基于重复的训练使 Twins 能够忽略噪声并专注于生物条件之间的差异。
他们测试了 Hi-C 标准化和测序深度的影响。在不同测序深度的数据集上进行训练后,Twins 可以学习测序深度伪影。然而,如果不同条件之间反映了测序深度的差异,则使用重复可以防止学习测序深度伪影。基于图像的 Twins 方法的一个局限性是它无法评估非常长范围的染色体内或染色体间相互作用。
Twins 可用于可靠的特征检测,并且学习到的卷积滤波器足以揭示因基因组组织者粘连蛋白和 CTCF 的扰动而产生的不同特征。研究人员设想 Twins 在染色质构象领域的许多其他应用,包括分析发育、稳态和再生中的正常状态与疾病状态。因此,Twins 与 Micro-C 和 Hi-C 的分析完全兼容非常重要。
总之,Twins 算法能够产生两个关键的有意义的输出。嵌入距离表明全基因组染色体位置条件之间的差异,并且可以与感兴趣的染色质特征相关联和比较。提取的差异特征表明变化的方向,以及特征的形状和大小。它们将作为选择合适工具进行染色体构象接触图下游定量的有用指南。
这项工作的一个主要成果是将染色质构象数据处理成一种可以应用于其他机器学习架构,从而构建该方法的范式。通过对比损失训练来利用重复可以与变化检测领域出现的全卷积网络等新方法相结合,以改变染色质构象数据的分析。
论文链接:https://www.nature.com/articles/s41467-023-40547-9
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










