将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

转换数 Kcat 是酶效率的衡量标准,对于理解细胞生理学和资源分配至关重要。由于绝大多数酶反应都无法进行实验 Kcat 估计,因此非常需要开发准确的计算预测方法。然而,现有的机器学习模型仅限于单一的、经过充分研究的生物体,或者除了与训练集中的蛋白质高度相似的酶之外,它们提供的预测不准确。
杜塞尔多夫大学(Heinrich Heine University,HHU)研究团队提出了 TurNuP,一种通用且独立于生物体的模型,可成功预测野生型酶自然反应的转换数。研究人员通过差异反应指纹来表示完整的化学反应,并通过经过修改和重新训练的蛋白质序列 Transformer Network 模型来表示酶,从而构建了模型输入。
该研究以「Turnover number predictions for kinetically uncharacterized enzymes using machine and deep learning」为题,于 2023 年 7 月 12 日发布在《Nature Communications》。

酶是所有活细胞中重要的生物催化剂。它们通常是大蛋白质,结合较小的分子(所谓的底物),然后将它们转化为其他分子,即「产物」。
如果没有酶,将底物转化为产物的反应就无法发生,或者只能以非常低的速率进行。大多数生物体拥有数千种不同的酶。酶在各种生物技术过程和日常生活中有着广泛的应用——从面包面团的发酵到洗涤剂。特定酶将其底物转化为产物的最大速度由所谓的转换数 Kcat 决定。
转换数 Kcat 是酶的一个活性位点将分子底物转化为产物的最大速率。Kcat 是酶活性定量研究的核心参数,对于理解细胞代谢、生理学和资源分配至关重要。特别是,综合的 Kcat 值对于考虑生产或维持酶的成本的代谢模型至关重要,这是准确模拟细胞生理学和生长的先决条件。
目前,还没有针对 Kcat 的高通量实验测定,并且实验既耗时又昂贵。因此,Kcat 估计对于大多数反应都是不可用的;即使对于大肠杆菌(可以说是生物化学上最具特征的生物体)来说,体外 Kcat 也仅占所有酶催化反应的约 10%。
在细胞代谢的基因组规模动力学模型中,这个问题通常通过采样缺失的 Kcat 值或将其拟合到大型数据集来解决。然而,这些技术通常会导致不准确的结果,并且拟合的 Kcat 值与已知的体外估计值几乎没有关系。
杜塞尔多夫大学的研究团队提出了一种通用机器和深度学习方法,用于预测野生型酶自然反应的体外 Kcat 值。与以前的方法相比,该团队通过考虑反应的整套底物和产物的数字指纹来表示化学反应。为了捕获酶的特性,研究人员使用经过微调的最先进的蛋白质表示作为额外的模型输入。
他们使用 Transformer Networks 创建了这些酶表示,这些网络经过数百万个蛋白质序列的训练。事实证明,对于各种预测任务,Transformer 网络的性能优于使用卷积神经网络(CNN)创建的蛋白质表示,后者在之前的模型中用于预测酶转换数。
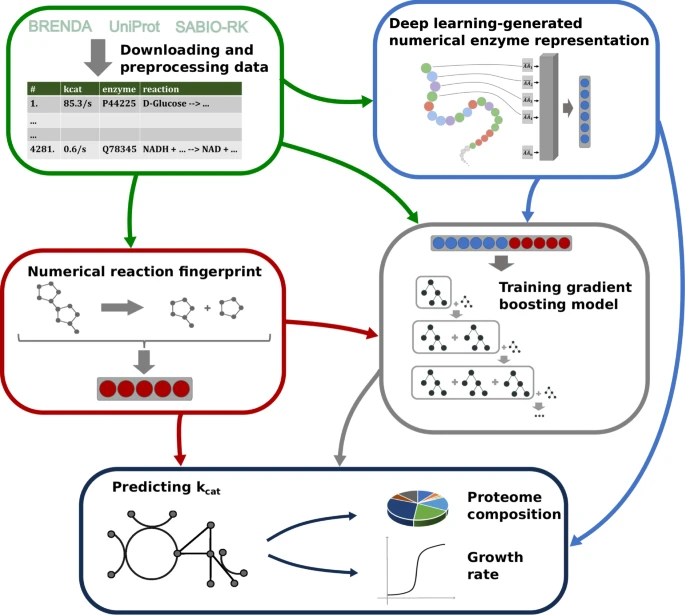
图示:机器学习模型根据数字酶表示和反应指纹来预测 Kcat。(来源:论文)
该团队构建的 Turnover Number Prediction model——TurNuP——优于简单的基于相似性的方法和之前预测 Kcat 的方法。测试表明,TurNuP 甚至可以很好地推广到与训练集中的蛋白质序列同一性小于 40% 的酶。
论文第一作者 Alexander Kroll 表示:「TurNuP 的性能优于之前的模型,甚至可以成功用于与训练数据集中相似度较低的酶。」除非至少 40% 的酶序列与训练集中的至少一种酶相同,否则之前的模型无法做出任何有意义的预测。相比之下,TurNuP 已经可以对最大序列同一性为 0 至 40% 的酶做出有意义的预测。
使用不同酵母物种的基因组规模、酶约束的代谢模型,研究人员证明使用 TurNuP Kcat 预测进行参数化可以改善蛋白质组分配预测。
通讯作者 Martin Lercher 教授补充道:「在我们的研究中,我们表明 TurNuP 所做的预测可用于比迄今为止的情况更准确地预测活细胞中酶的浓度。」
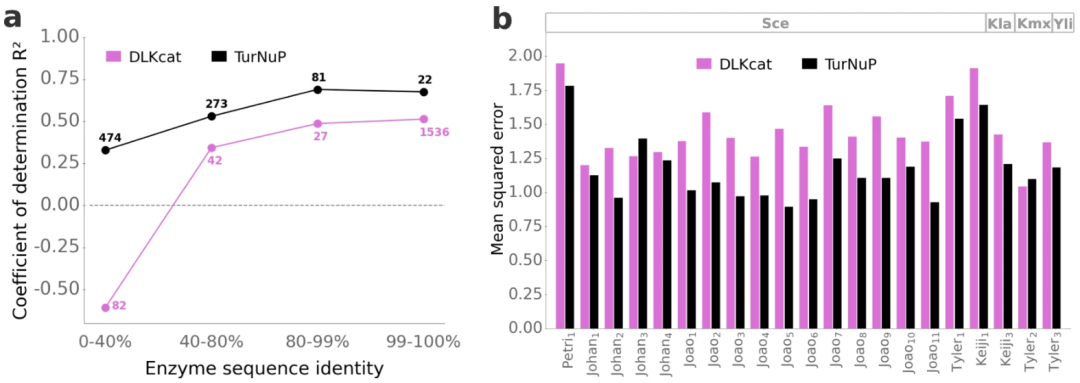
图示:TurNuP 对于训练集中类似于蛋白质的酶的预测更加准确,并且优于现有的深度学习模型。(来源:论文)为了促进 TurNuP 模型的广泛使用,该团队不仅为生物信息学家提供了用于大规模 Kcat 计算的 Python 函数,而且还构建了一个易于使用的网络服务器,无需专门的软件。
服务器地址:https://turnup.cs.hhu.de
论文链接:https://www.nature.com/articles/s41467-023-39840-4
相关报道:https://phys.org/news/2023-07-ai-tool-enzymes.html
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。










