此篇文章来自 GEN Biotechnology 的 2022 年 8 月月刊内容「Deep Learning Concepts and Applications for Synthetic Biology」。本文概述了与合成生物学相关的数据类别和深度学习架构,强调了利用深度学习在合成生物学中实现新的理解和设计的新兴研究,并讨论了这一领域的挑战和未来机会。再创对此综述进行了中文翻译和整理,并为读者带来第一部分内容,主要涉及一些合成生物学与深度学习的基本概念。
William A.V. Beardall, Guy-Bart Stan, and Mary J. Dunlop / 作者
锐锐 / 翻译
合成生物学与深度学习具有天然的协同效应。合成生物学可以生成大规模的数据集来训练模型,例如通过 DNA 合成;深度学习模型可以用于指导设计,比如生成新的元件或给出是最佳实验的建议。对于目前能够生成大规模高质量数据集的合成生物学领域,新兴的研究结果展示了将深度学习与工程生物学相结合的潜力,包括设计新型生物元件、蛋白质结构预测、显微镜数据的自动分析、最佳实验设计和生物分子实现人工神经网络等成功案例。
科研人员的目标通常是开发一个深度学习模型,这个模型在接受许多典型示例的训练后,能够推广到对未曾见过的输入进行输出预测。深度学习模型的一个关键特征是通过在人工神经网络的各层之间逐步传递信息,这些模型可以逐步从输入数据中提取信息。例如,在分析显微镜图像时,网络的早期层可能提取水平或垂直边缘等低层特征,而后续层则综合这些信息来识别图像中的细胞模式或形状。此外,深度学习网络可以编码输入值之间的复杂非线性关系。例如,从蛋白质序列预测蛋白质功能的深度学习模型可以学习到某些氨基酸的特定组合具有协同作用,提高活性,超出了各个氨基酸贡献所预期的水平。
为了推动合成生物学和深度学习交叉领域的未来发展,我们需要克服几个挑战。从教育培训的角度来看,合成生物学家通常没有接受过传统的深度学习方法的教育,难以同时跟上这两个快速发展的领域。此外,合成生物学数据集具有特定领域的限制。在某些领域存在大量数据,例如自然序列信息,但这些数据集的多样性有限,因为包含的非功能性序列或导致非常高表达的序列通常较少。相反,由于合成生物学构建在实现和测试方面的实际限制,其他应用在可用数据量方面受到严重限制。
本综述旨在帮助合成生物学家了解和利用深度学习方法在其研究中的应用,提供方法概述,并总结工程生物学和深度学习交叉领域的最新研究进展。主要分为以下五个板块:
描述与合成生物学相关的数据类别,并解释如何在数学上表示它们,以便作为深度学习模型的输入。
介绍最新的深度学习在合成生物学中的应用进展,重点介绍元件设计、基于结构的学习、成像和其他领域的示例。
回顾近期工程生物学中将深度学习网络应用于生物分子的研究工作。
一个经过适当训练的深度学习网络可以输入数据并准确预测输出。输入数据通常被表示为数字的矩阵或向量。这些数学表示对于将生物学问题转化为适合模型训练的问题至关重要。确定特定问题的最佳数据表示对于开发高性能和可推广的模型至关重要,因为表示方式决定了向模型输入的信息,并限制了可应用的学习算法集合。
我们必须谨慎选择数据表示方式,以确保问题相关的自变量得到恰当表示,同时限制不相关或混淆的变量数量——模型必须学会忽略这些变量。巧妙选择数据表示方式可以提高数据效率。在这里,我们描述了合成生物学相关数据的常见类型以及它们如何以数字形式表示。
由于测序能力的快速发展,我们在序列组合空间方面拥有大量的数据。这些数据可以包括 DNA、RNA 或氨基酸序列。通常,这些数据使用嵌入式 (embeddings) 表示为矩阵,或者使用将序列元素映射为向量的函数。最基本的嵌入方法是独热编码 (one-hot encoding),因为在每个嵌入向量中只有一个元素是“hot”的,取值为 1,而其他所有元素都为 0。例如,一个由核酸字符串表示的序列(例如 ATTGGTCA)会被转换为一个矩阵,其中「行」表示可能的值,如 A、T、G 或 C,「列」表示序列中的位置(图 1A)。
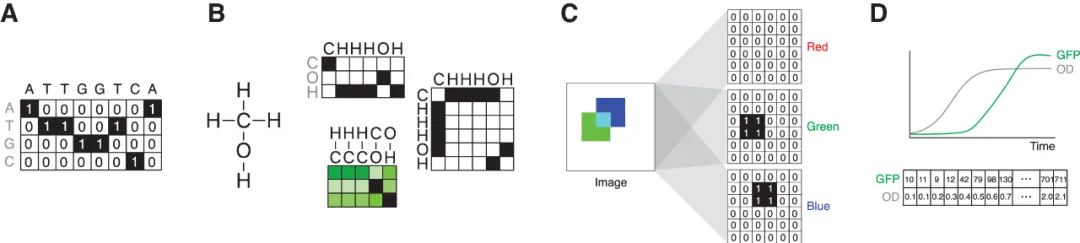
▲ 图1:与合成生物学相关的数据类别及其数学表示。
(A) 序列数据的 one-hot encoding。
(B) 以节点(原子)特征、边(键)特征和节点邻接矩阵编码的图结构分子数据。
(C) 以每个颜色通道的矩阵表示的图像。
(D) 时间序列数据。GFP,绿色荧光蛋白;OD,光密度。
因此,一个长度为 50 的序列可以用一个 4×50 的矩阵来表示。同样地,蛋白质序列数据可以使用每个氨基酸的独热编码来表示,因此一个 300 个氨基酸的序列可以表示为一个 20×300 的矩阵。独热编码很直观,但在某些情况下可能会限制模型的表征能力,因为它忽略了在序列的某个位置上的某些氨基酸可能表现出相似的特性,例如亲疏水性。从大规模未标记的蛋白质数据集中学习到的氨基酸嵌入表示法,已被证明在某些蛋白质工程任务中优于独热编码方法。
序列表示法可以单独使用,也可以与附加的生物物理特征结合使用。例如,在一个启动子计算模型中,La Fleur 等人使用了 -35 和 -10 位点的独热编码的组合,以及对序列不同部分的能量贡献相对应的特征作为模型的输入。
无论是小分子还是大分子,分子的结构都可以通过多种方式几何描述,可以是基于字符串的表示,如 SMILES 或其衍生物,或者学习嵌入(Learned embedding)表示。另外,分子可以通过其结构式来表示,并将其结构式编码为图形(图 1 B),在此基础上可以直接应用基于图形的学习方法。图中的节点是分子中的原子,一组节点特征定义了原子的标识和属性,如原子质量和电荷。边可以定义为分子中的原子之间的化学键,可选地包含边的特征和边的权重。
例如,边的特征可能包括化学键的类型,边可以根据键长进行加权。这样,分子的结构式可以完全定义为一组节点特征、一组边特征和一个邻接矩阵,该矩阵编码了节点之间的连接关系。这种方法的优势在于明确地将重要概念(如原子的局部性)纳入学习框架中,并将分子几何结构作为算法的约束条件,这在药物发现领域近年来取得了显著的进展。
另一方面,分子可以被视为三维空间中的对象,通过为其组成的原子赋予明确的坐标以及现有的节点特征。这些坐标和节点特征可以在机器学习工作流中使用。请注意,这些概念可以抽象为分子结构的更高层次视图,例如,将节点定义为 DNA 和 RNA 结构中的核苷酸,以及蛋白质中的氨基酸。
合成生物学实验也可以生成图像数据,例如显微镜图像文件。在这种情况下,像素在矩阵的行和列中表示,矩阵中的数值对应图像中的灰度值(图 1 C)。如果图像包含多个颜色通道,则维度会扩展以包括这些值。例如,一个大小为 400×600 像素的彩色图像由一个 400×600×3 的对象表示,其中包含与红、绿和蓝色通道相关联的数据。
时间序列数据,例如来自酶标仪的读数,也可以输入到深度学习网络中。在这种情况下,数据点被表示为一个数字向量,向量中的每个条目对应特定时间点的值(图 1 D)。也可以将此扩展为包括多种类型的数据。例如,对于 100 个时间点测量的绿色荧光蛋白表达和光密度的读板仪数据可以表示为一个 2×100 的矩阵。
更一般地说,合成生物学应用可以生成各种“组学”数据集,例如基因组学、转录组学、蛋白质组学或表观遗传学数据,可以使用分类编码或与生化特性相关的数值表示,比如前文提到的独热编码。
人工神经网络(Artificial Neural Networks,ANNs)是一种受生物神经网络启发的机器学习算法。在 ANNs 中,人工神经元是用于模拟生物神经元行为的数学函数。ANN 模型用于识别模式、分类数据和执行其他特定任务。深度学习是机器学习的一个子领域,它使用具有多层人工神经元的网络来学习数据中的复杂模式。
深度学习拥有众多的架构,在这篇综述中,我们介绍了一些目前在合成生物学应用中常用的表示方法。其中包括传统的深度学习网络,如多层感知器(multilayer perceptrons),卷积神经网络(CNNs)在计算机视觉领域被广泛使用,也在序列分析中证明了其有效性,以及考虑信息顺序或重
要性的网络,如循环神经网络(RNNs)和 Transformer。
我们还讨论了适用于利用生物化学结构的几何性质的网络,例如图神经网络方法。需要注意的是,这并不是一个全面的建模方法列表。例如,强化学习和生成模型等技术在合成生物学中也具有巨大的潜力。除了网络架构外,模型的训练、验证和测试也是关键考虑因素,这些因素对性能至关重要。附录中简要介绍了这些概念,并提供了更详细内容的引用。(再创的读者们可点击查看原文进行浏览)
传统的 ANN 架构使用一组“神经元”,每个神经元接收一系列的数值输入。这些输入乘以称为权重的参数,并加上一个称为偏差的常数。然后,这个数经过非线性函数处理,成为神经元的输出(图 2 A)。在历史上,研究人员使用 S 型函数作为非线性函数,但现代深度学习网络的大多数实现在隐藏层中的神经元使用修正线性单元(ReLU)作为非线性函数,这是出于计算效率的考虑。通常一个网络中有许多神经元,相同的输入乘以每个神经元的不同权重。例如,如果输入对应于 DNA 序列信息,则权重可以调整每个核苷酸如何决定最终的输出(例如转录活性)。在输入是多维数组的情况下,它可以展开为向量(例如,将一个 4×50 矩阵展开成一个 200 维的向量)。
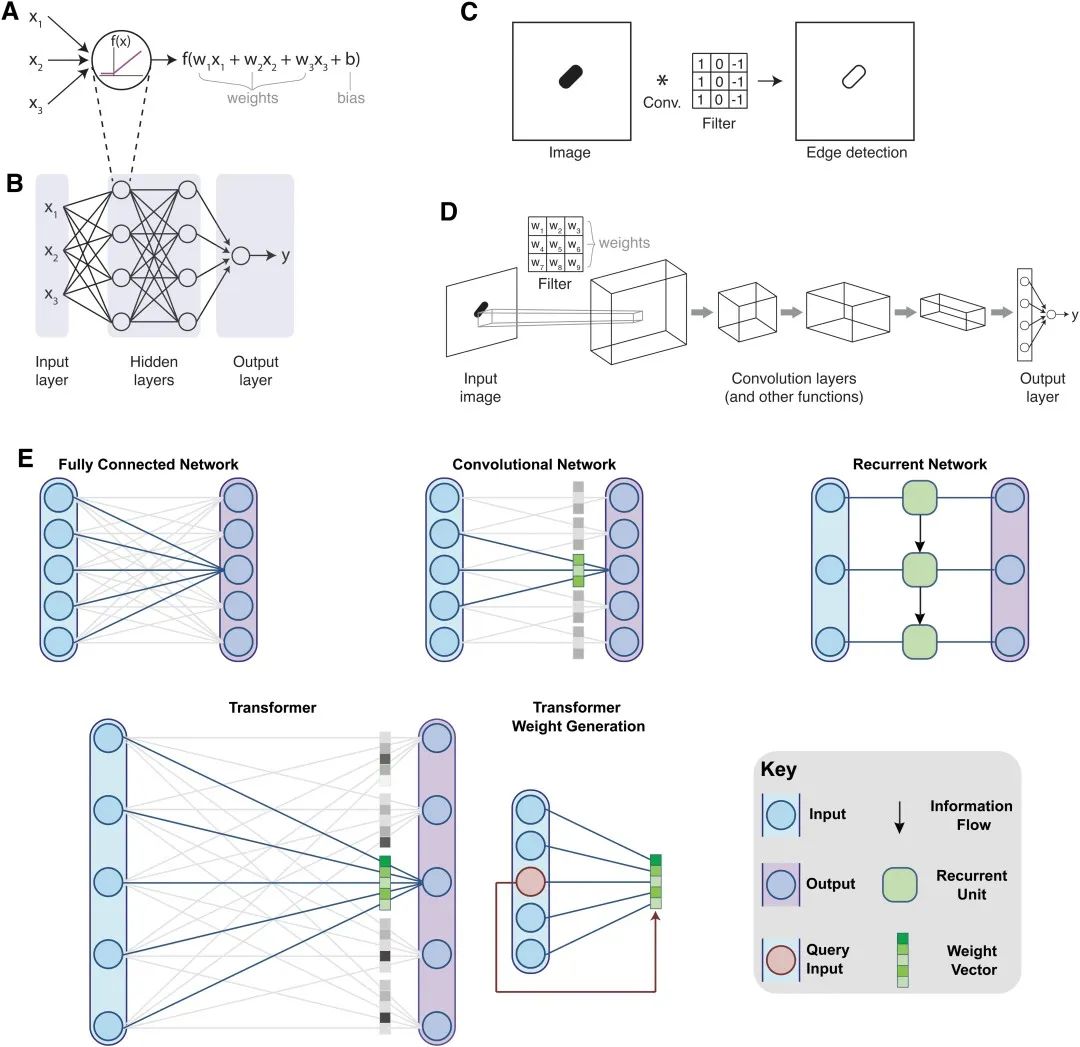
▲ 图2:深度学习网络架构。
(A) 单个人工神经元接收输入,输入×权重+偏差项。然后,这个值被传入非线性函数(如图中所示的 ReLU 函数)进行处理。
(B) 多层感知器网络由多个隐藏层组成,每个神经元与其周围层中的神经元完全连接。
(C) 卷积操作将一个滤波器应用于数据,例如图像。滤波器中的值决定了卷积实施的操作,如边缘检测。图中展示了一个代表性的用于从亮到暗的转变中检测垂直边缘的滤波器示例,将该滤波器应用于具有这种亮-暗模式的图像区域将产生一个较大的值。可以与其他滤波器结合使用以检测边缘。
(D) 卷积神经网络(CNNs)使用多个顺序的卷积滤波器,通常与其他操作结合使用,其中权重是滤波器中的值。
(E) 不同网络架构层中信息流的比较。除非另有说明,信息从左向右流动。在全连接网络中,例如在多层感知器中,每个输入都与每个输出相连接,每个连接都有一个学习的权重(为简单起见,未显示权重)。卷积层只在其局部区域内将输出与输入相连接。连接在一组输出中共享权重,作为应用于整个输入的滤波器。RNNs逐个处理输入的每个元素,将信息从每次迭代传递到下一次。Transformers 是全连接的,但其权重由输入数据生成。对于每个输出,从相应的输入生成查询(红色),将其与每个输入进行比较以确定对每个输入的注意力。CNN,卷积神经网络;ReLU,修正线性单元;RNN,循环神经网络。
卷积神经网络(Convolutional Neural Networks,CNNs)可以保留「附近的数据如何相对排列」的局部位置信息。此外,它们使用参数共享结构,在整个输入中应用相同的模型权重。
在传统的基于滤波器的分析任务中,滤波器中的数值是手动选择的,用于指定用户认为重要的属性,例如边缘检测(图 2 C)。相反,CNNs 将滤波器参数作为模型的权重,这些权重由网络学习得来
(图 2 D)。CNNs 通常使用一系列卷积步骤来执行顺序分析操作,这些操作可以抽象出特征,例如模式或颜色梯度。卷积层通常穿插在执行其他数学运算的层之间,例如池化,用于通过降低数据的维度来集中信息。CNNs 还可以包含其他网络架构的元素,例如在卷积层后面跟随全连接层。
循环神经网络(Recurrent Neural Networks,RNNs)是一类专为序列数据设计的模型。它们通过迭代处理数据序列,并根据内部状态和输入序列的下一个值来递归更新模型的内部状态(或记忆)(图 2 E)。传统上,这些网络常用于语言处理,其中单词的顺序对于上下文和意义至关重要。同样,这些网络适用于处理生物学的时间序列数据或序列信息。例如,在处理 DNA 序列时,起始和终止密码子的相对位置对于确定蛋白质表达非常重要。然而,这些网络的递归性质存在一些限制。最重要的是,简单的 RNNs 由于梯度消失问题无法学习远距离元素之间的长期依赖关系,并且它们的递归性质禁止并行实现,限制了其可扩展性。
随着长短期记忆(LSTM)网络的发展(Long Short-Term Memory,LSTM),RNNs 的性能得到了关键的提升。LSTM 模型旨在通过为模型添加长期记忆状态来改善 RNNs 的有限时间记忆能力,在长期记忆在某些领域中,模型必须做出明确的决策,以添加到或删除信息到长期记忆中。例如,如果模型试图预测给定 mRNA 是否会翻译成蛋白质,终止密码子的存在可能会被放入长期记忆,并一直保留,直到识别出下游的起始密码子为止。
Transformer 是一种较新的用于序列数据的模型,通过跟踪序列数据中的关系(如这句话中的单词)来学习上下文并因此学习含义。它消除了 RNN 变体所遇到的内存有限问题,同时通过消除递归实现了更高的计算效率和可并行化。Transformer 在基于序列的任务上表现出具有开创性的,改变范式的性能,全面超越了 RNNs 和 LSTMs。它甚至在计算机视觉问题上展现了超越 CNNs 的能力,尽管它最初并不是为这些任务设计的。Transformer 避开了模型记忆的概念,允许模型一次性查看并生成整个数据序列中每个位置的输出,从而实现了这种革命性的性能。
对于每个输出,模型会选择从序列的哪些部分中获取信息。这是通过所谓的 “注意力” 机制实现的,其中模型可以学习在序列的每个点上哪些信息是重要的,并专注于传递这些信息(图 2 E)。例如,预测可能形成二级结构的小 RNA 行为的模型,它可能会更专注于对与感兴趣序列互补的序列上(例如,对于 “CGA” 的输出将包含很多来自包含 “UCG” 的序列的信息)。
序列和图像数据的学习方法利用了数据的规则欧几里得结构和它所赋予的空间局部性的自然概念。其他结构化数据,例如分子的结构式图、DNA 和 RNA 的二级结构图,或蛋白质的原子坐标数据,则不具有这些相同的结构属性。然而,它们具有自己的对称性和局部性定义,这些特性可以用来构建学习框架。图神经网络是专为将欧氏神经网络中的信息流推广到图结构而设计的,通过节点之间不规则的边连接来编码结构的局部性,定义了一种可扩展和可推广的节点之间传递信息的方法。
这使得我们可以对结构化数据的高质量表示进行学习,执行节点标记或边缘预测任务,或跨结构汇集在一起,并输入到多层感知器中,在分子尺度上执行分类或回归。
(未完待续。)











