2022年11月18日,中南大学李敏教授团队在Briefings in Bioinformatics上发表文章RLBind: a deep learning method to predict RNA–ligand binding sites。在本文中,作者提出了一种深度学习模型RLBind,通过结合全局RNA序列通道和局部相邻核苷酸通道,从序列依赖性和结构依赖性特性预测RNA-小分子结合位点。这项研究首次开发了用于RNA-小分子结合位点预测的卷积神经网络。实验结果表明,RLBind在预测结合位点方面优于其他最先进的方法,表明RNA中全长序列的全局信息和有限的局部相邻核苷酸的局部信息的结合可以提高模型对结合位点预测的预测性能。RNA-小分子结合位点的鉴定在RNA靶向药物的发现和开发中起着至关重要的作用。与靶向蛋白质的常规疗法相比,这些小分子有望成为指导新型RNA靶向疗法开发的主要化合物。RNA可以提供许多具有不同结构和功能的潜在药物靶点。然而,到目前为止,只有少数方法被提出。预测RNA-小分子结合位点仍然是一个巨大的挑战。需要新的计算模型来更好地提取特征并更准确地预测RNA-小分子结合位点。
在这项研究中,作者通过整合源自全长和局部相邻RNA序列和结构特性的全局和局部特征,构建了一个基于深度卷积神经网络的模型,以预测RNA-配体结合位点(图1)。作者使用全长RNA和局部相邻RNA基序来计算输入特性,即序列相关特性和结构相关特性,被组合在一起作为10D矢量编码的输入。更具体地说,序列特性包括核苷酸类型、进化保守性评分(evolutionary conservation)。结构性质包括网络拓扑性质、生物化学性质和可及表面积(accessible surface area,ASA)。将输入特性输入到卷积神经网络和密集神经网络,分别提取全长RNA序列的全局特征和有限相邻核苷酸的局部特征。
在局部特征的构造中,滑动窗口大小(2 m + 1) 用于提取核苷酸的局部特征,其中目标核苷酸每侧的m个邻居被认为是相关的邻居信息。这里,m=5,滑动窗口的大小是11。当一个核苷酸在右窗口或左窗口中没有足够的邻居时,会填充相同数量的零以匹配缺失的长度。RNA序列具有不同的长度。因此,在构建全局特征时,定义了L=64的固定长度来创建有效的表示形式。根据基准数据集中的长度分布,固定长度可以覆盖至少80%的RNA。对于比固定长度长的RNA序列,该研究将其截断,并将较短的RNA填充为0。考虑到配体结合确实可以发生在RNA分子的截短部分,使用64的固定长度会使一些信息丢失。然而,只有<20%的RNA在截短部分中存在结合残基。与最大长度相比,固定长度的使用将减少模型参数。随后,通过核大小为17的卷积层处理具有固定长度的RNA序列。接下来,最大池化层的输出和局部特征被连接并馈送到两个密集层中。在这里,作者使用了两个具有192个和96个节点的密集层。最后,应用具有sigmoid激活函数的输出层来预测作为结合位点残基的概率。
每个核苷酸具有10维的特征。序列依赖性特性包括四维的独热编码的核苷酸类型(C,U,G,A)和一维编码的进化保守性。结构相关属性方面,RNA的结构在深入了解RNA的功能和进化过程中起着至关重要的作用。因此,本研究采用了三种类型的结构依赖性性质,即二维编码的网络拓扑性质、二维编码的生物化学性质(包括分子量和侧链pKa)和一维载体编码的ASA。在RNA三级结构网络中,一个高度接近的核苷酸几乎位于网络的中心。同时,高度核苷酸通常形成局部结合腔。作者使用RNA三级结构构建了网络,其中核苷酸被定义为节点,两个非连续核苷酸的一对重原子之间的非共价相互作用被定义为边缘。更具体地说,设置了8埃的距离阈值来获得网络边缘。然后,计算两个网络拓扑性质,即节点的接近中心性(closeness)和度(degree),以评估节点的重要性。
作者将RLBind与多种方法进行对比。使用两个测试集Test18和Test9来评估不同计算方法的性能。如图2所示,RLBind的性能优于其他比较方法。例如,计算了MCC(马修斯相关系数)。对于Test18数据集,RLBind显示出比RNAsite(0.253)、RBind(0.187)、Rsite(0.071)和Rsite2(0.01)高得多的MCC(0.324)。同样,对于Test9数据集,RLBind(0.546)也绝对优于RNAsite(0.426)、RBind(0.287)、Rsite(0.159)和Rsite2(0.072)。此外,RLBind在Test18数据集上的AUC比RNAsite相对更低,但在Test9数据集上比其他方法的AUC更高。精确度和召回率是评估结合位点预测准确性的重要指标。RLBind的表现与RNAsite相似,但在Test9组中优于RBind、Rsite和Rsite2。RLBind对Test18集合的精度结果也优于其他比较方法。此外,RLBind在两个测试集上都显示出显著更高的召回率。在本研究中,各种序列和结构输入特性是互补的,在特征提取中起着至关重要的作用。在这里,作者进行了一项消融研究,以测试不同性质的重要性。如表1所示,网络拓扑性质对结合位点的识别有显著影响。从生物学的角度来看,紧密度可以识别长程变构效应的RNA结构网络的中枢,而程度可以识别短距离相互作用的配体结合腔。此外,ASA在结合位点识别中也发挥着重要作用。事实上,配体倾向于嵌入RNA结构的内部,而位于RNA结构内部的核苷酸通常具有较低的ASA。另一方面,分析表明,序列和结构特性的结合可以提高RNA-配体结合位点预测的性能。
表1 消融实验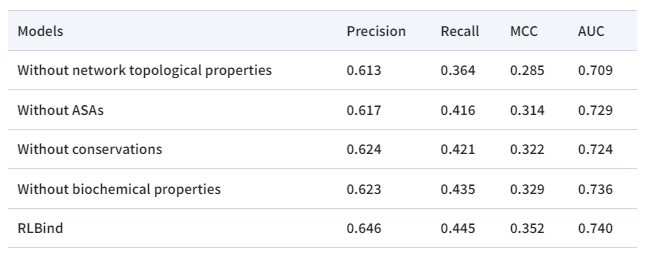 为了进一步评估RLBind在预测中的效率,作者选择了几种RNA进行案例研究。靶向RNA以设计治疗疾病的新型化合物显示出巨大的潜力。例如,tau外显子10剪接调控元件(PDB代码:1EI2)形成一个主要凹槽,与药物样小分子(新霉素)结合,这与人类额颞叶痴呆和帕金森病有关。RNA 1EI2的结合位点包括13个核苷酸,RLBind可以成功识别8个结合位点残基,没有假阳性(图3A)。RNA 1EI2的精密度为1.0,召回率为0.615。前Q1核糖开关适体(PDB代码:3Q50)是一种重要的调节元件,可以通过与小分子配体结合来促进或减弱基因表达。更具体地说,RNA 3Q50的结合位点由八个核苷酸组成,令人兴奋的是,可以成功鉴定出七个结合位点残基(图3B)。RNA 3Q50的精密度为1.0,召回率为0.875。图3是tau外显子10剪接调控元件(PDB编码:1EI2)结合配体NMY(A)和前Q1核糖开关适体(PDB代码:3Q50)结合配体PRF(B)的表面表示。预测的和未预测的结合表面分别用红色和蓝色表示。结合位点的高预测准确性可能有助于设计靶向这些结合位点的新药来治疗一些特定的疾病。
为了进一步评估RLBind在预测中的效率,作者选择了几种RNA进行案例研究。靶向RNA以设计治疗疾病的新型化合物显示出巨大的潜力。例如,tau外显子10剪接调控元件(PDB代码:1EI2)形成一个主要凹槽,与药物样小分子(新霉素)结合,这与人类额颞叶痴呆和帕金森病有关。RNA 1EI2的结合位点包括13个核苷酸,RLBind可以成功识别8个结合位点残基,没有假阳性(图3A)。RNA 1EI2的精密度为1.0,召回率为0.615。前Q1核糖开关适体(PDB代码:3Q50)是一种重要的调节元件,可以通过与小分子配体结合来促进或减弱基因表达。更具体地说,RNA 3Q50的结合位点由八个核苷酸组成,令人兴奋的是,可以成功鉴定出七个结合位点残基(图3B)。RNA 3Q50的精密度为1.0,召回率为0.875。图3是tau外显子10剪接调控元件(PDB编码:1EI2)结合配体NMY(A)和前Q1核糖开关适体(PDB代码:3Q50)结合配体PRF(B)的表面表示。预测的和未预测的结合表面分别用红色和蓝色表示。结合位点的高预测准确性可能有助于设计靶向这些结合位点的新药来治疗一些特定的疾病。此外,作者分析了测试集中的预测结果,发现配体在RNA结构中的位置可能会影响预测的准确性。例如,小分子通过识别RNA的浅表面空腔与RNA 1DDY(维生素B12 RNA适体)相互作用。RNA 1DDY有八个结合位点残基,RLBind只能识别三个结合位点的残基和六个假阳性(图4A)。RNA 1DDY的准确度为0.333,召回率为0.375。较低的预测精度表明,当小分子位于RNA的浅表面空腔时,很难识别结合位点。事实上,小分子倾向于结合在生物分子的更深空腔中,该空腔具有更大的静电势、库仑吸引或氢相互作用。RNA 1NEM(一种RNA适体)由16个结合位点残基组成;RLBind可以识别10个结合位点残基(图4B)。预测精度为1.0,召回率为0.625。更高的预测精度表明,当分子埋在更深的RNA结构腔中时,可能更容易识别结合位置。图4中,预测的和未预测的结合位点分别用红色和蓝色表示。假阳性显示为青色。
RNA中对于小分子结合位点预测的发展可以为揭示RNA配体识别的机制和设计药物发现中的新化合物提供基础。然而,目前用于结合位点预测的方法的准确性仍然需要提高。基于结合特异性,RLBind是一种整合局部和全局特征的深度学习方法,用于预测RNA中的小分子结合位点。此外,基于序列的特征和基于结构的特征被认为分别有助于提取序列进化特性和结构拓扑信息。在这里,这些输入特征被输入到深度学习模型中,以预测来自全局和局部模式通道的小分子结合位点。更具体地说,RLBind训练局部基序和全局RNA来推断结合位点。与其他计算方法相比,RLBind具有更好的预测性能。
事实上,RNA中的小分子结合位点识别可以促进药物设计的发展。特别是,具有更多预测真阳性和更少预测假阳性的模型将加速这一过程。此外,研究表明,形状匹配、静电相互作用、疏水相互作用和氢相互作用也在小分子结合识别中发挥着重要作用。在未来的研究中,预测任务中将考虑这些特征。总之,作者开发了一个基于深度学习的模型RLBind,用于预测RNA中的小分子结合位点。研究表明,将全长RNA的全局信息和RNA中有限相邻核苷酸的局部信息相结合,可以提高该模型对RNA-小分子结合位点预测的预测性能。
参考文献
[1] Wang et al. RLBind: a deep learning method to predict RNA–ligand binding sites. Brief Bioinform. 2023
感兴趣的读者,可以添加小邦微信(zhiyaobang2020)加入读者实名讨论微信群。添加时请主动注明姓名-企业-职位/岗位 或 姓名-学校-职务/研究方向。









