大家好,感谢大家对王初课题组一直以来的关注和支持。在此,小编祝大家新年快乐,兔年吉祥。机器学习如今已经渗透到化学生物学研究的方方面面,今天为大家总结一下2022年期间王初课题组公众号推送的机器学习相关的文章,供各位读者查阅。
2021年,以AlphaFold2和RoseTTAFold为代表的深度学习结构预测方法获评Nature Methods年度方法,对单体蛋白质的结构预测达到了前所未有的精度,且正在不断启发更多的下游应用。
我们知道,预测蛋白与其它蛋白/多肽的相互作用、蛋白质上的各种修饰、金属离子等配体结合对结构的影响也将非常有价值。例如,【Nat. Commun. | 利用蛋白质折叠神经网络进行多肽-蛋白对接】一文中,作者通过多聚甘氨酸链连接需要对接的蛋白和多肽序列,随后使用AlphaFold2结构预测方法直接预测多肽-蛋白复合物结构,发现只需对运行参数进行少量优化,AlphaFold2即可达到与专门处理多肽-蛋白对接任务的PIPER-FlexPepDock相当的精度,且具有很快的运行速度。作者还特别指出,AlphaFold2能够预测多肽结合时诱导的蛋白构象变化,而这是传统的生物物理算法难以实现的。
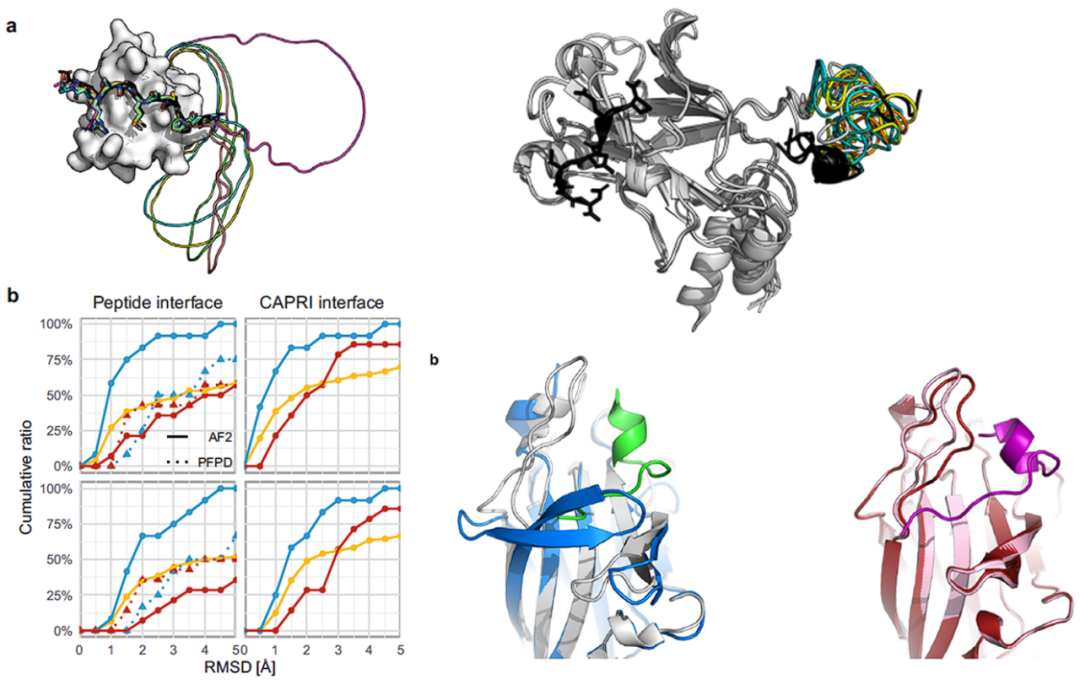
【专题 | 蛋白质和多肽相互作用】一文中详细介绍了更多将机器学习用于蛋白-多肽复合物体系上的进展,例如【Improving Peptide-Protein Docking with AlphaFold-Multimer using Forced Sampling】文中,作者将DeepMind团队专门为复合物优化的Alphafold-multimer用于多肽-蛋白复合物结构预测问题中,并指出在网络推理过程中也启用dropout可采样更广阔的结构空间,进一步提升多肽-蛋白复合物预测精度。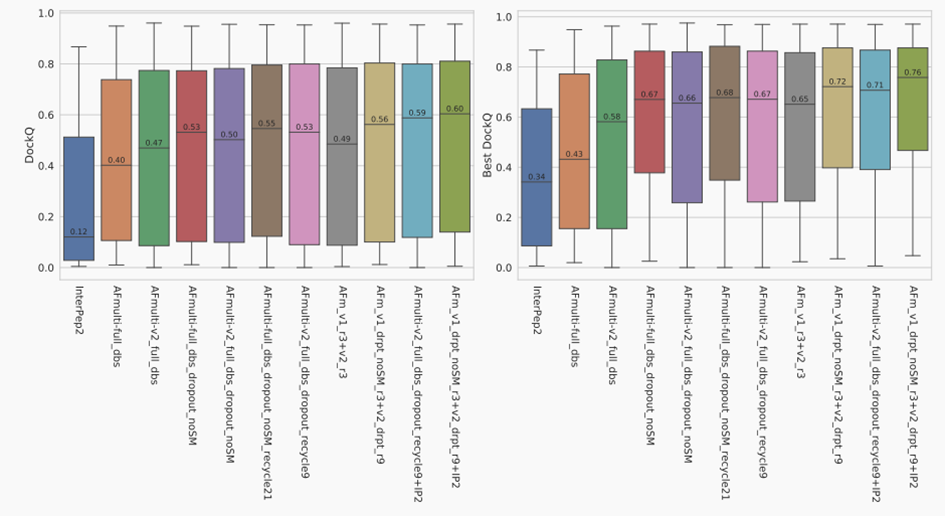
尽管AlphaFold2在单蛋白结构预测上取得了巨大成功,然而其对抗体、特别是抗体互补决定区域(CDR)预测的准确性还很不理想。由于抗体高质量晶体结构的稀缺,通过预训练的蛋白质语言模型提取序列表征成为抗体预测的一种主流策略。【Patterns | 可解释的深度学习模型预测抗体结构】一文中,作者提出DeepAb模型,通过预训练语言模型使用双向长短期记忆网络(Bi-LSTM)对抗体轻链和重链序列进行逐残基编码,转化为固定长度的隐藏状态,传递到使用长短期记忆网络(LSTM)的解码器进行解码。此外,抗体序列还并行地通过1D ResNet,并与预训练语言模型的输出进行组合输入到2D ResNet中,2D ResNet是结构预测模块的主要组件。值得注意的是,此前的蛋白结构预测模型往往是不可解释的,无法提供结构生物学上的见解。本文作者将2D ResNet的输出通过交叉注意力层转化到六个输出分支,分别代表Fv结构中的关键参数(距离、平面角和二面角),最后这些输出将被作为几何约束应用到Rosetta优化中。最终模型在表现上优于基准方法,并具有一定的可解释性。
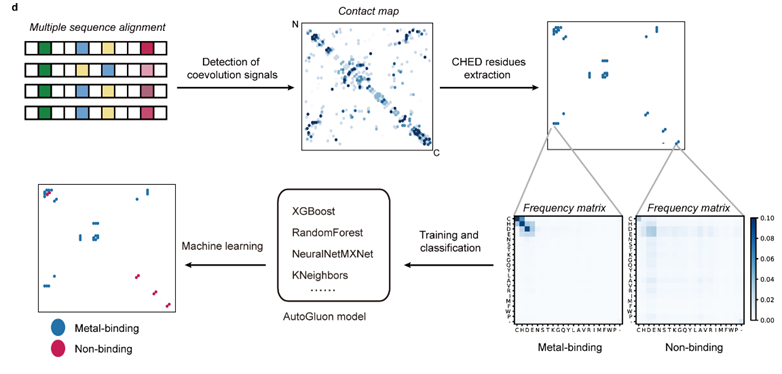
 蛋白质功能的正确行使不仅仅依赖于其自身的折叠结构,同时也依赖于其经历的各种结合和修饰事件,而这些信息目前在AlphaFold2方法中是缺失的。例如,金属离子具有独特的物理和化学性质,因而经常被用来稳定蛋白质结构、参与物质运输、作为蛋白质中的辅助因子来帮助催化生化反应和信号传导等等,在许多生物过程中起着不可或缺的作用。据估计,整个蛋白质组中超过三分之一的蛋白是金属结合蛋白。在【号外 | 王初课题组与苏晓东课题组合作发展基于共进化和机器学习的蛋白质金属结合位点预测新方法】一文中,开发了一种名为MetalNet的计算方法。作者发现共进化信号在金属结合位点中高度富集,基于此现象从多序列比对中计算残基之间的共进化信号,以此训练机器学习模型,实现了金属结合蛋白和金属结合位点的高置信度预测,并通过实验验证了大肠杆菌中的新型金属结合蛋白citX,为研究金属蛋白质组和金属生物学提供了新的工具。
蛋白质功能的正确行使不仅仅依赖于其自身的折叠结构,同时也依赖于其经历的各种结合和修饰事件,而这些信息目前在AlphaFold2方法中是缺失的。例如,金属离子具有独特的物理和化学性质,因而经常被用来稳定蛋白质结构、参与物质运输、作为蛋白质中的辅助因子来帮助催化生化反应和信号传导等等,在许多生物过程中起着不可或缺的作用。据估计,整个蛋白质组中超过三分之一的蛋白是金属结合蛋白。在【号外 | 王初课题组与苏晓东课题组合作发展基于共进化和机器学习的蛋白质金属结合位点预测新方法】一文中,开发了一种名为MetalNet的计算方法。作者发现共进化信号在金属结合位点中高度富集,基于此现象从多序列比对中计算残基之间的共进化信号,以此训练机器学习模型,实现了金属结合蛋白和金属结合位点的高置信度预测,并通过实验验证了大肠杆菌中的新型金属结合蛋白citX,为研究金属蛋白质组和金属生物学提供了新的工具。
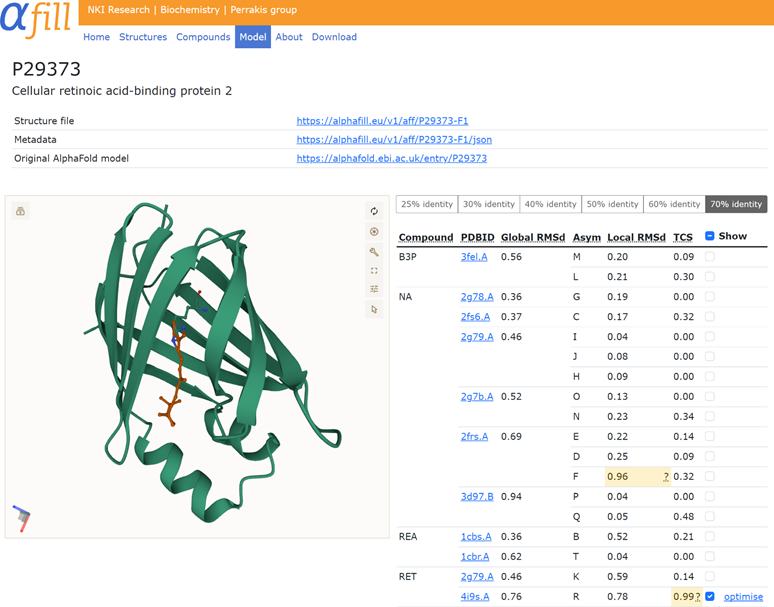
 无独有偶,【Nat. Methods | AlphaFill:使用配体信息丰富AlphaFold2模型】一文中也指出配体信息的重要性,作者开发了AlphaFill方法,用于将各种配体信息引入AlphaFold2模型中。他们对于AlphaFold数据库中的每一个蛋白,在PDB-REDO数据库中寻找同源结构。从同源性最强的序列开始,对于包含特定配体的蛋白质结构,经过与AlphaFold2模型的全局和局部两次结构比对(局部结构比对给出local RMSD值),将配体分子“移植”至AlphaFold2模型中。如果配体含有不同的构象,则分别地被记录于AlphaFill模型中。作者计算了AlphaFill模型与实验晶体结构中配体局部的RMSD(定义为LEV打分),发现LEV与local RMSD相关性很好,验证了配体坐标“移植”的有效性。同时还评估了配体和蛋白质原子之间可能的冲突(定义为TCS打分),结果表明LEV与TCS密切相关。针对不同的local RMSD值,作者给出了相应的置信度用于参考,最终实现了非聚合物配体向AlphaFold2模型的引入。
无独有偶,【Nat. Methods | AlphaFill:使用配体信息丰富AlphaFold2模型】一文中也指出配体信息的重要性,作者开发了AlphaFill方法,用于将各种配体信息引入AlphaFold2模型中。他们对于AlphaFold数据库中的每一个蛋白,在PDB-REDO数据库中寻找同源结构。从同源性最强的序列开始,对于包含特定配体的蛋白质结构,经过与AlphaFold2模型的全局和局部两次结构比对(局部结构比对给出local RMSD值),将配体分子“移植”至AlphaFold2模型中。如果配体含有不同的构象,则分别地被记录于AlphaFill模型中。作者计算了AlphaFill模型与实验晶体结构中配体局部的RMSD(定义为LEV打分),发现LEV与local RMSD相关性很好,验证了配体坐标“移植”的有效性。同时还评估了配体和蛋白质原子之间可能的冲突(定义为TCS打分),结果表明LEV与TCS密切相关。针对不同的local RMSD值,作者给出了相应的置信度用于参考,最终实现了非聚合物配体向AlphaFold2模型的引入。
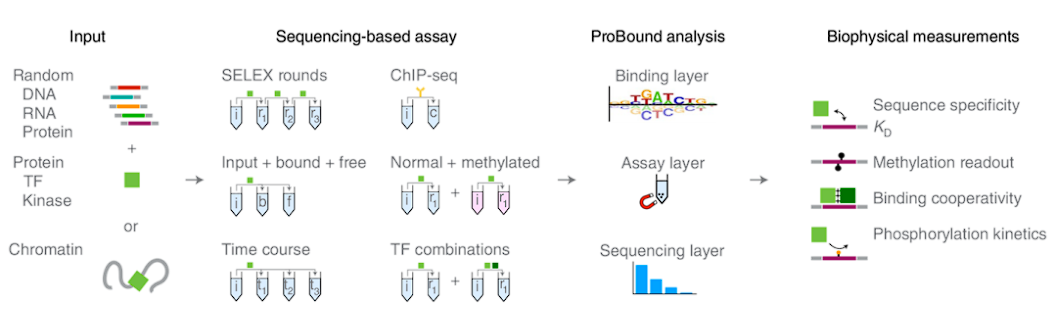
除金属和小分子配体外,基因调控和信号转导等关键的细胞过程依赖于序列特异性的分子识别,以引导蛋白优先与特定的核酸或多肽配体相互作用。【Nat. Biotechnol. | 利用可解释机器学习从测序数据中预测蛋白质配体结合亲和力】一文中指出,这种序列识别的强度和特异性通常跨越几个数量级,甚至弱配体也能发挥功能。大规模并行测序大大提高了序列识别的速度,特别是高通量测序方法与随机配体库的体外选择相结合,已成为无偏分子相互作用分析的有力工具。这包括用于转录因子(TF)和RNA结合蛋白的SELEX方法,以及用于蛋白酶和T细胞受体的蛋白展示方法。作者开发的灵活机器学习框架ProBound,通过Binding、Assay、Sequencing三个层组合成一个似然函数,在大量测序数据下进行训练和优化,实现了蛋白-配体结合过程中多个生物物理学参数可解释地预测。
酶的改造与设计一直是蛋白设计领域最为困难的问题之一。【Nature|机器学习辅助设计解聚PET的水解酶】一文中指出,PET水解酶即使经过广泛的工程化改造,其热稳定性和催化活性仍不理想。作者认为高度集中的蛋白质工程方法不能考虑整体稳定性和活性之间的权衡,而中立的、基于结构的深度学习神经网络通常可以改善酶功能。为此,作者使用三维自监督卷积神经网络(CNN)MutCompute算法来识别稳定性突变。该算法学习氨基酸残基的局部化学微环境,在来自蛋白质数据库(PDB)的19,000多个序列平衡的蛋白质结构上进行训练,可以轻松预测蛋白质野生型(WT)中氨基酸没有优化到最佳的位置。通过对预测突变进行实验组合和验证,他们最终得到的FAST-PETase与野生型PETase相比包含五个突变,在30到50°C和一系列pH值之间显示出相对于野生型和工程替代品来说优异的PET水解活性。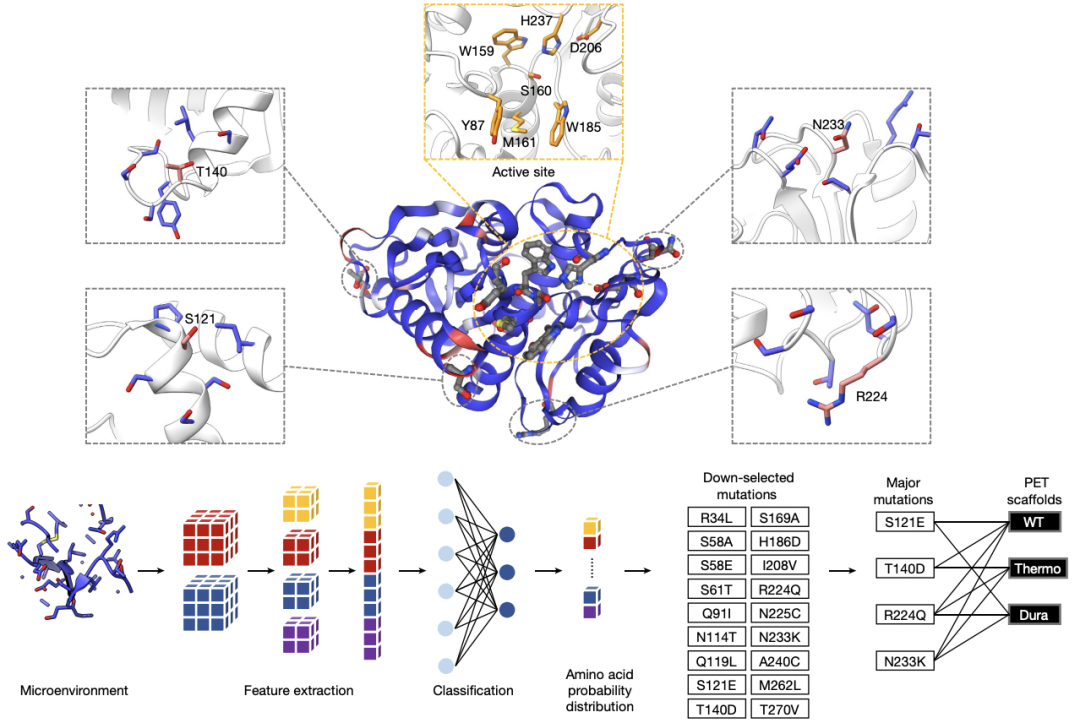
除蛋白质外,机器学习还被广泛运用于化学生物学关注的其它功能分子。例如,顺式作用元件的突变可以使基因的表达发生改变、进而改变生物的表型和适应性。构建顺式作用元件DNA序列到表达量的映射关系,是生物学中一个长期存在的挑战。【Nature | 非编码DNA调控序列的进化,演化性和工程】一文中,作者对80bp的DNA序列进行随机采样,将80bp的DNA序列插入质粒中黄色荧光蛋白基因的启动子中,随后将质粒导入酵母中进行表达。以此法测量了三千万条启动子DNA序列和它们在复合培养基下酵母中的表达水平,两千万条启动子DNA序列和它们在尿苷缺乏培养基下酵母中的表达水平。随后用这两类信息进行训练,获得了具有优秀预测性能的深度神经网络模型,能够正确预测表达工程中DNA序列与表达量的关系。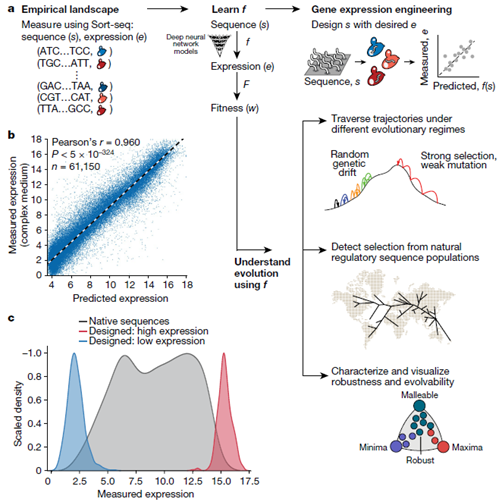
RNA上存在大量的修饰类型,其中最常见的RNA修饰之一是m6A修饰,这种修饰主要发生在DRACH(D-A/G/U,R-A/G, H-A/C/U)这一motif上,该修饰已经被报道会影响RNA结构、稳定性、剪接和翻译。【Nat. Methods | 使用多实例学习框架从直接RNA测序中检测m6A】一文中,作者指出从实验中获得的带修饰的训练标签是在位点水平上为一组reads提供,而不是为每个单独的read提供训练标签,这被称为多实例学习(MIL)问题。作者开发了名为m6Anet的方法,基于MIL的神经网络模型,它采用信号强度和序列特征两方面的信息来从RNA-Seq 数据中识别潜在的m6A修饰位点。他们的模型考虑了修饰和未修饰 RNA 的混合,并输出训练数据中所有DRACH 5-mer在任何给定位点的 m6A 修饰概率。与现有方法不同,m6Anet从每个候选位点学习单个read的高维表示,然后将它们聚合在一起以产生更准确的m6A位点预测。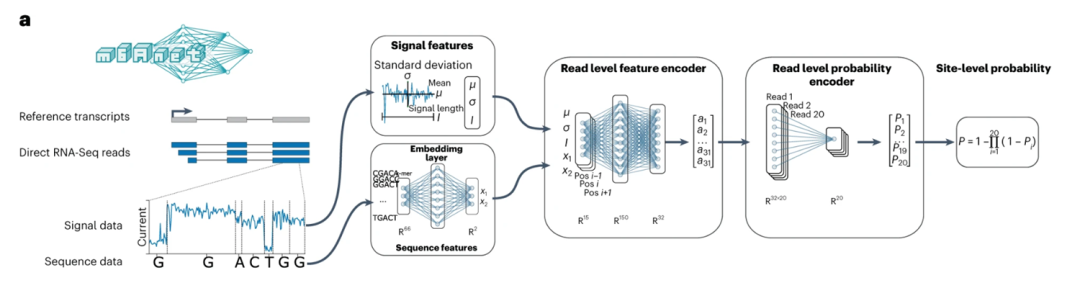
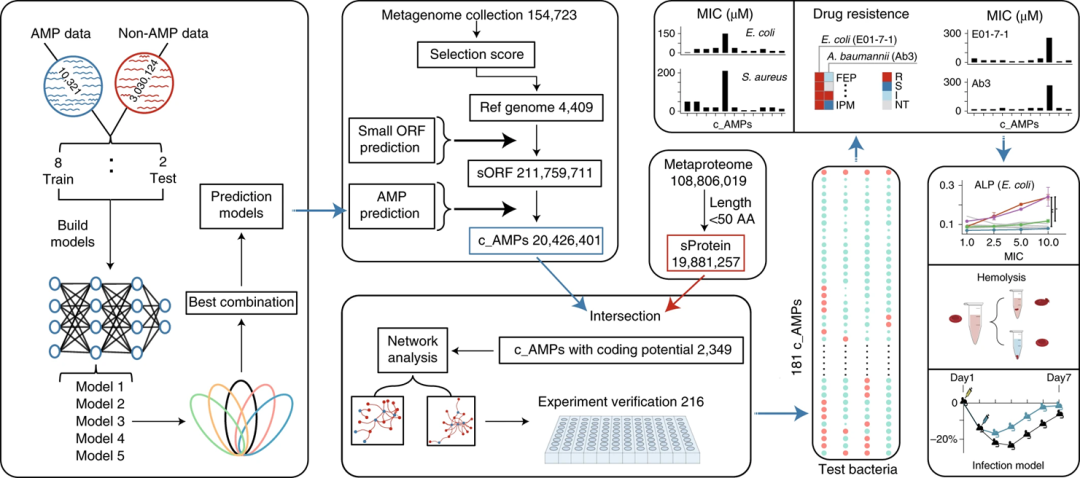
现有的许多抗生素和其他药物都来源于微生物代谢物。在具有生物活性的次级代谢产物中,短肽因其具有高度的多样性和广泛的生物活性而受到人们关注,尤其是来自细菌的抗菌肽 (AMP)。【Nat. Biotechnol. | 使用深度学习从人类肠道微生物组中鉴定抗菌肽】一文中,作者综合了多种语言模型网络,包括LSTM、Attention和BERT,形成了一个统一的管道,用于从人类肠道微生物组数据中识别候选 AMP。通过对宏基因组群中开放阅读框序列进行预测,作者确定2,349个候选 AMP,通过化学合成了候选 AMP 中的216个,其中181个显示出抗菌活性(阳性率 > 83%),这些肽中大多数与训练集中的 AMP 的序列同源性低于40%,且展示出显著的抗菌功效。
PROTACs 是一种双功能小分子,它的一头能够与 E3 泛素连接酶结合,另一头可以与靶蛋白结合。它的两头通过一段 linker 连接。当 E3 泛素连接酶-PROTACs-靶蛋白三者形成复合物以后,E3 连接酶给靶蛋白打上泛素化标签,随后,该泛素化的靶蛋白被蛋白酶体所识别和降解。【Nat. Commun. | 基于深度学习的PROTACs预测器DeepPROTACs】一文中指出,对于 PROTACs 小分子来说,其 linker 对于靶蛋白的降解能力是非常重要的,但是一个linker是否有效合适,目前只能够通过大量的实验试错进行验证。作者借助图卷积神经网络算法,可以根据POI和E3连接酶的结构以及PROTAC分子的结构作为输入,有效地预测给定PROTACs的降解功效,首次尝试了用深度学习技术助力PROTACs小分子的开发和设计。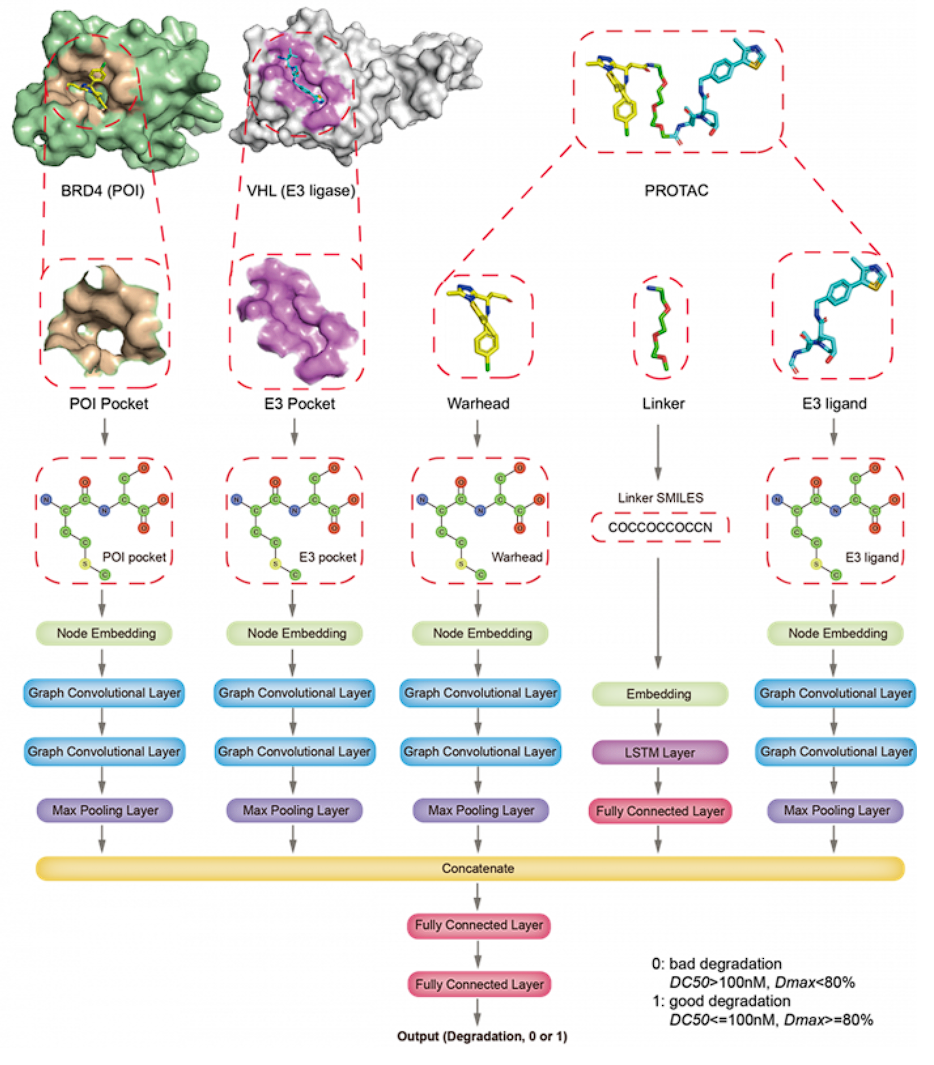
以上为王初课题组2022年分享的文献中有关机器学习的主要内容,欢迎大家交流与学习。










