继药物研发机器学习平台 TorchDrug 之后,时隔一年,Mila 唐建团队开源了新的蛋白质机器学习平台 TorchProtein,这是目前第一个专门针对蛋白质研究的开源机器学习库。
蛋白质是生物体的重要组成成分。理解蛋白质的结构与生化性质,对于药物研发和人类健康有着不可估量的意义。传统基于生物实验的蛋白质研究不仅周期漫长,而且开销巨大。相比之下,机器学习技术则能大幅降低蛋白质研究的周期和开销,为新药的研发带来革命性的影响。然而,基于机器学习的蛋白质研究,涉及到生物领域知识、机器学习算法、并行实现等多个方面,具有较高的入门门槛。市面上也缺少合适的开源库来支持这方面的研究,致使机器学习技术在蛋白质研究中发展受阻。近日,Mila 唐建团队联合英伟达、英特尔、IBM 以及蛋白质设计初创公司百奥几何共同开源了蛋白质机器学习平台 TorchProtein。TorchProtein 在此前开源平台 TorchDrug 的基础上,为蛋白质打造了一套专用的模块组件。TorchProtein 不仅提供了处理蛋白质的数据结构、主流的算法模型,还包括了标准数据集和任务评测接口。其所有接口均有很强的可扩展性,满足各类机器学习算法开发的需要。无论是图机器学习、蛋白质语言模型还是自监督训练,都能轻松基于 TorchProtein 实现。
唐建教授表示,未来机器学习辅助下的蛋白质研发依赖于丰富开源社区的培养。「我们期待这个平台能成为未来机器学习蛋白质研发主要的开源平台,并推动这一方面的进展。」唐建说道。考虑到不同任务需要用到蛋白质分子、序列和结构等不同信息,TorchProtein 设计了一套统一不同模态信息的数据结构。可对蛋白质进行分子、序列或结构层面的操作,并在模态之间进行无缝切换。平台提供了多种基于蛋白质序列与结构的模型,仅需一两行代码即可调用 TorchProtein 中的标准模型来分析蛋白质序列和结构数据。针对在蛋白质结构上构图繁琐的问题,TorchProtein 还专门提供了灵活的即时构图的模块,支持在 GPU 上动态构图。TorchProtein 中引入了大量蛋白质数据集和相关基准测试任务,并记录了主流的机器学习算法在这些测试任务上的测试结果,为新的算法研究提供代码与实验支持。针对基于蛋白质序列和结构的预测任务,平台提供了许多大规模预训练模型,这些模型将有效促进蛋白质机器学习在实际中的运用,并大大缩减计算成本。
下面我们将从数据结构到算法模型,依次介绍 TorchProtein 的功能:TorchProtein 使用统一的图数据结构来表征蛋白质序列和结构,是对 TorchDrug 中图数据结构的特化。我们可以通过蛋白质结构 PDB 文件来构建数据,并指定使用原子、氨基酸残基和化学键特征。
from torchdrug import data, utilsfrom rdkit import Chemimport nglview
pdb_file = utils.download("https://files.rcsb.org/download/2LWZ.pdb", "./")mol = Chem.MolFromPDBFile("2LWZ.pdb")view = nglview.show_rdkit(mol)view
protein = data.Protein.from_pdb(pdb_file, atom_feature="position", bond_feature="length", residue_feature="symbol")print(protein)
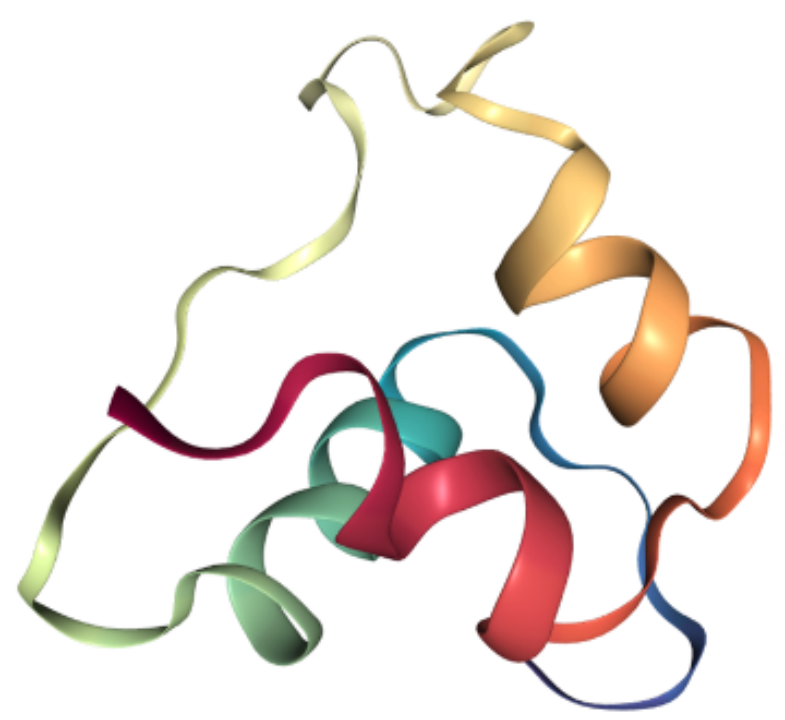
Protein(num_atom=445, num_bond=916, num_residue=57)利用这一数据结构,我们可以轻松获取蛋白质序列信息,TorchProtein 同样支持从蛋白质序列来构建数据结构。aa_seq = protein.to_sequence()print(aa_seq)seq_protein = data.Protein.from_sequence(aa_seq, atom_feature="symbol", bond_feature="length", residue_feature="symbol")print(seq_protein)
FVNQHLCGSDLVEALYLVCGERGFFYTDPTGGGPRRGIVEQCCHSICSLYQLENYCNProtein(num_atom=445, num_bond=910, num_residue=57)
为了充分利用 GPU 资源,TorchProtein 支持将多个蛋白质打包处理,且数据可以在 CPU 和 GPU 之间自由切换。
proteins = [protein] * 3proteins = data.Protein.pack(proteins)proteins = proteins.cuda()print(proteins)
PackedProtein(batch_size=3, num_atoms=[445, 445, 445], num_bonds=[916, 916, 916], num_residues=[57, 57, 57], device='cuda:0')
与 PyTorch 中的张量类似,TorchProtein 的蛋白质数据结构支持按照氨基酸残基的位置进行索引、切分和重组。
segments = [protein[:2], protein[2:4], protein[4:6], protein[6:8]]segments = data.Protein.pack(segments)segments.visualize()
TorchProtein 也提供了动态建图、蛋白质序列和结构分割、原子级别和氨基酸级别结构切换等功能,详细操作指南可参考官网教程。
TorchProtein 提供了许多用于蛋白质序列性质分析的数据集、任务和模型,尽可能避免重复编写代码,帮助用户快速评估各模型在各数据集上的性能。以蛋白质在细胞中位置预测数据集(Subcellular Localization)为例,我们可以通过两行代码构建数据集并获取其训练集、验证集和测试集。
from torchdrug import datasets
dataset = datasets.SubcellularLocalization("~/protein-datasets/", residue_only=True)train_set, valid_set, test_set = dataset.split()
接着,我们可以定义一个简单的两层 CNN 模型作为蛋白质序列编码器,并在这个 CNN 基础上定义任务模型来进行蛋白质细胞位置预测。
from torchdrug import core, models, tasks
model = models.ProteinCNN(input_dim=21, hidden_dims=[1024, 1024], kernel_size=5, padding=2, readout="max")task = tasks.PropertyPrediction(model, task=dataset.tasks, criterion="ce", metric=("acc", "mcc"), num_mlp_layer=2)
TorchProtein 提供了多功能的求解器(solver)来执行模型训练、评测和模型参数保存。
import torch
optimizer = torch.optim.Adam(task.parameters(), lr=1e-4)solver = core.Engine(task, train_set, valid_set, test_set,optimizer, batch_size=64, gpus=[0])solver.train(num_epoch=100)solver.evaluate("valid")solver.save("subloc_cnn.pth")
在 TorchProtein 平台上,研发团队评测了各蛋白质序列机器学习模型在各基准任务上性能,在蛋白质细胞位置预测基准上的结果如下。详细基准测试结果见 PEER Benchmark 论文(https://arxiv.org/abs/2206.02096)。
蛋白质结构性质分析
TorchProtein 也提供了多种用于蛋白质结构性质分析的数据集和模型,以供用户进行模型评估,进而促进蛋白质结构分析的实际应用。以 Enzyme Commission(EC)蛋白质功能预测数据集为例,通过两行代码我们可以轻松构建数据集并获取其训练集、验证集和测试集。dataset = datasets.EnzymeCommission("~/protein-datasets/")train_set, valid_set, test_set = dataset.split()
TorchProtein 提供了各种蛋白质结构编码器的实现,这里我们选择使用当前最优之一的 GearNet-Edge 作为蛋白质结构编码器,并在此基础上构建任务模型以解决 EC 数据集中多个二分类问题。
model = models.GearNet(input_dim=21, hidden_dims=[512, 512, 512, 512, 512, 512], num_relation=7, edge_input_dim=59, num_angle_bin=8, batch_norm=True, concat_hidden=True, short_cut=True, readout="sum")task = tasks.MultipleBinaryClassification(model, num_mlp_layer=3, criterion="bce",task=[_ for _ in range(len(dataset.tasks))], metric=["auprc@micro", "f1_max"])
同样,我们可以利用 TorchProtein 的多功能求解器来进行模型训练、评测和模型参数保存。
import torch
optimizer = torch.optim.Adam(task.parameters(), lr=1e-4)solver = core.Engine(task, train_set, valid_set, test_set,optimizer, batch_size=4, gpus=[0])solver.train(num_epoch=100)solver.evaluate("valid")solver.save("ec_gearnet_edge.pth")
TorchProtein 团队在 EC 和 GO 两个蛋白质功能预测基准上评测了各种蛋白质结构编码模型的性能,结果如下。实现和评测细节见 GearNet 论文(https://arxiv.org/abs/2203.06125)。
这一平台的项目负责人为加拿大蒙特利尔学习算法研究所(Mila)副教授、终身教授唐建,其研究领域包括几何深度学习、图表示学习、图神经网络、药物发现及知识图谱。唐建 2014 年毕业于北京大学信息科学技术学院并获得博士学位,2014-2016 年任职微软亚洲研究院副研究员,2016-2017 年为密歇根大学和卡内基梅隆大学的联合培养博士后,曾获 2014 年机器学习顶级会议 ICML 的最佳论文。唐建是图表示学习领域的代表性学者,他所提出的网络表示学习算法 LINE 被广泛应用,其他代表工作还包括 RotatE 等。Mila 实验室是由深度学习先驱 Yoshua Bengio 教授领导的人工智能实验室(https://mila.quebec/),主要从事深度学习、强化学习、优化算法等人工智能领域的基础研究以及在不同领域的应用。TorchProtein 整个项目由博士生张作柏、徐明皓、朱兆成、袁新钰,以及多位来自蛋白质设计初创公司百奥几何、IBM 研究院、英特尔和英伟达的工业界合作者,以及多位来自剑桥大学、清华大学和北京大学的实习生共同完成。
感兴趣的读者,可以添加小邦微信(zhiyaobang2020)加入读者实名讨论微信群。添加时请主动注明姓名-企业-职位/岗位 或
姓名-学校-职务/研究方向。









