——背景介绍——
蛋白通过与DNA相互作用来执行一系列的功能,如转录、基因表达调控、基因剪切等。目前的一些实验结构数据提供了很好的依据以增强人们对蛋白质分子构象发生转变的理解,包括氢键、氨基酸理化性质、静电相互作用、范德华相互作用等。一方面,测序技术的发展使得这几年序列数据呈指数增长,由于实验技术有限,目前只有少部分的复合物数据能够使用,藉此,研究者尝试发展计算方法来预测DNA结合残基位点。这些方法大致分成了3类,一种是基于序列的,另一种是基于结构的,还有一种是混合以上2种方式。这些算法可以在表1中找到。由于预测精度远得不到满足,来自Indraprastha Institute of Information Technology的Patiyal等人在Briefings in Bioinformatics发表了一种基于深度学习的算法来提高DNA结合残基位点预测的精度,并且该算法只用到了序列的数据,模型框架也非常简单。——方法原理——
数据集:作者从已经发表的两篇文献hybridNAP和ProNA2020中分别收集到了864和308个注释的蛋白序列。为了去冗余,作者在数据集上用了CD-HIT,其中要求测试集的数据和训练集之间的序列一致性要低于30%。随后作者利用构建的模型在以上两种方法使用的训练和测试数据进行重新训练和测试。最后他们创建了一个包含646个蛋白序列的训练集,和46个蛋白序列的独立测试集。最后训练集中总共包含15636个有相互作用的残基,和298503个没有相互作用的残基,测试集中包含965个有相互作用的残基和9911个没有相互作用的残基。
模式尺寸:选取中心残基左右两边各8个,或者9个(针对两端残基)构建序列环境,具体的流程图见图1;
图1 特征生成(A)和模型发展(B)的流程图。图(A)中(b)是二元谱,(c)是物理化学特征谱成分分析:考虑到不同残基结合的偏好性,作者归纳了25个明显不同的物理化学特征成分。模式谱:二元谱将21种残基(20+X)用0-1热编码。所以对于一个长度为17的片段,总共有17*21=357个特征,类似地物化谱有25*17=425个特征,PSSM谱有21*17=357个特征。
将以上的特征整合到标准的1DCNN网络中(图2),就可以做模型训练了。训练的时候为了避免过拟合,作者用了5折交叉验证。
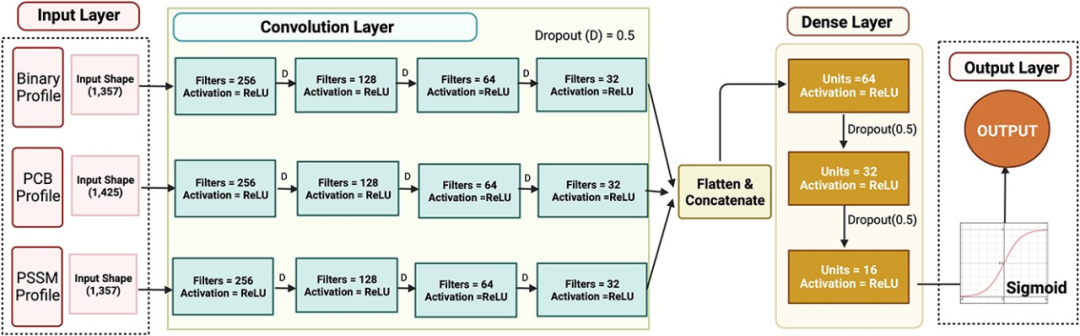
——结果——
作者分析了不同氨基酸残基相互作用和非相互作用残基的百分比(图3),也计算了不同残基结合DNA的倾向性(图4)。可以看到,K,R,W和Y是最受欢迎的结合位点,而疏水残基整体表现都很不好。
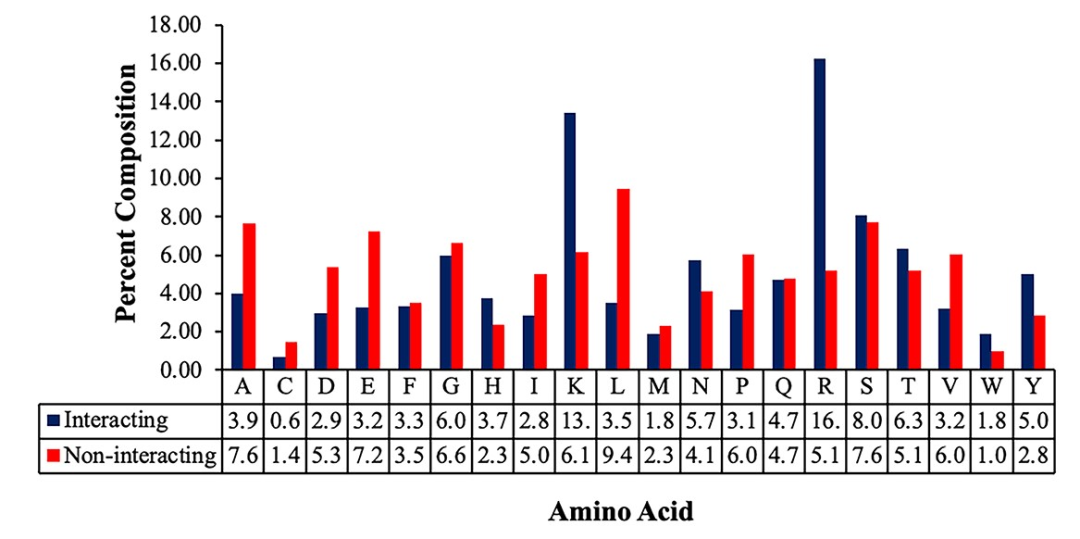
图3 相互作用和非相互作用残基的百分比
图4 标准化的残基DNA的倾向性
随后作者依次将三种特征谱分别训练,得到三个结果,分别如表2,表3,和表4所示。
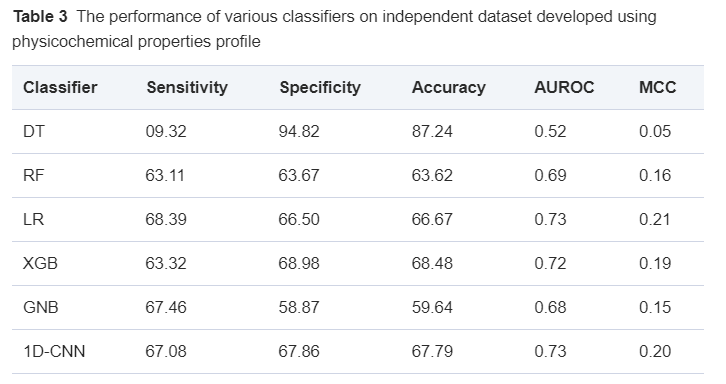
表3 物理化学谱特征得到的结果
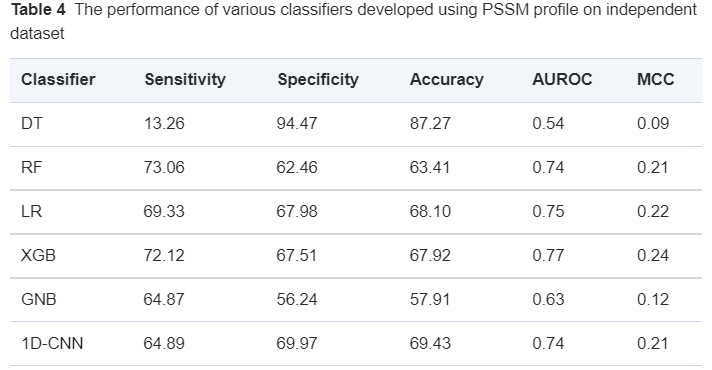 表4 PSSM谱得到的特征表2-4中的DT代表决策树,RF是随机森林,LR是逻辑斯蒂回归,XGB是极端梯度提升,GNB是高斯朴素贝叶斯。可以看到,相比于二元谱特征和物理化学谱特征,PSSM谱提供的信息更多,AUROC也最大。随后作者将这几类特征进行整合,得到结果如表5所示,发现AUROC和MCC都有进一步的增大,并且1D-CNN的效果最佳。
表4 PSSM谱得到的特征表2-4中的DT代表决策树,RF是随机森林,LR是逻辑斯蒂回归,XGB是极端梯度提升,GNB是高斯朴素贝叶斯。可以看到,相比于二元谱特征和物理化学谱特征,PSSM谱提供的信息更多,AUROC也最大。随后作者将这几类特征进行整合,得到结果如表5所示,发现AUROC和MCC都有进一步的增大,并且1D-CNN的效果最佳。表5 整合特征结果
接下来,作者跟其他的方法进行比较,如表6中所显示的,作者的方法DBPred取得了最高的AUROC和MCC。
表6 不同方法的性能比较分析
本文采用了大量的不同的机器学习技术,并且发现基于PSSM谱特征的方法对结果贡献最多。作者发展的1D-CNN的方法达到了最高水平。
——感悟——
随着越来越先进和复杂的深度学习模型出现,模型的表现能力得到进一步增强,但是有时候一些简单模型也能达到很不错的效果,参数优化也更快更容易收敛。总之,能解决问题的都是好模型。
[1] Patiyal
S, Dhall A, Raghava G P S. A deep learning-based method for the prediction of
DNA interacting residues in a protein[J]. Briefings in Bioinformatics, 2022.[2] https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbac322/6658239?login=true
点击左下角的"阅读原文"即可查看原文章。
本文为GoDesign原创编译,如需转载,请在公众号后台留言。






