本质上,inferCNVpy这个包是InferCNV的python版重现。主要还是遵循R包版本的计算步骤,进行了少量修改。inferCNVpy通过使用numpy、scipy和稀疏矩阵,使其计算效率大大提高。inferCNVpy可以在Linux,Mac环境下运行。Windows下可参考:
inferCNVpy github见:https://github.com/icbi-lab/infercnvpy
官网认为这个方法仍处于实验阶段,但他们还是决定将其放到GitHub上,因为它可能会有用。但是需要大家小心看待结果:
首先是inferCNV软件的环境配置:
conda create -n infercnv
conda activate infercnv
pip install infercnvpy
umap-learn 0.5.2版本有一点bug,如果下面的UMAP图像显示异常,可以考虑用pip做一个降级
在Linux环境下利用Jupyter Notebook编译器运行下述python代码:
import scanpy as sc
import infercnvpy as cnv
import matplotlib.pyplot as plt
sc.settings.set_figure_params(dpi=80,figsize=(5, 5))
Step1.载入示例数据
input的数据应该是已过滤后的单细胞数据,例如我们可以从Seurat流程进行QC过滤,然后导出数据。
示例数据是Smart-seq2生成的3000个单细胞:
adata = cnv.datasets.maynard2020_3k()
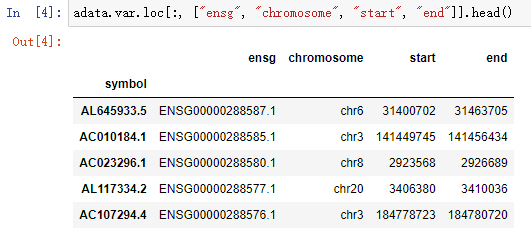
adata.var.loc[:, ["ensg", "chromosome", "start", "end"]].head()
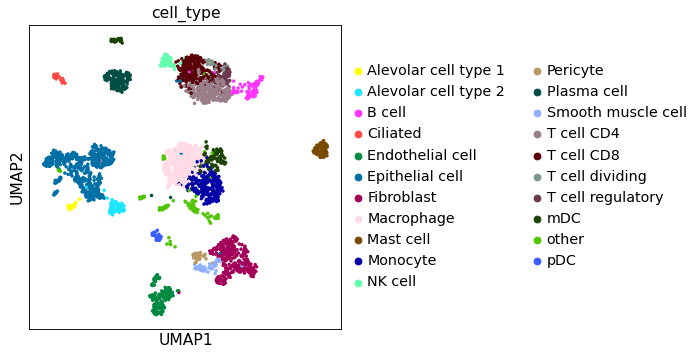
可视化:
sc.pl.umap(adata, color="cell_type")
Step2.Running infercnv
现在运行 infercnvpy.tl.infercnv()。本质上,该方法通过染色体和基因组位置对基因进行分类,并将基因组区域的平均基因表达与参考进行比较。原始的 inferCNV 方法使用 100 的窗口大小,但更大的窗口大小可能有意义,具体取决于数据集中的基因数量。
# We provide all immune cell types as "normal cells".
cnv.tl.infercnv(
adata,
reference_key="cell_type",
reference_cat=[
"B cell",
"Macrophage",
"Mast cell",
"Monocyte",
"NK cell",
"Plasma cell",
"T cell CD4",
"T cell CD8",
"T cell regulatory",
"mDC",
"pDC",
],
window_size=250,
)
3000个Smart-seq2单细胞约运行32.39秒。
与inferCNV R版教程一样,inferCNVpy可设置参考细胞:
- 最常见的用例是将肿瘤与正常细胞进行比较。如果有关于哪些细胞正常的先验信息(例如,来自基于转录组学数据的细胞类型注释),建议将此信息提供给 infercnv()。
- 可以提供的不同细胞类型越多越好。一些细胞类型在生理上过度表达某些基因组区域(例如浆细胞高度表达基因组相邻的免疫球蛋白基因)。如果提供多种细胞类型,则仅考虑与所有提供的细胞类型不同的区域受 CNV 影响。
- 如果不提供任何参考,则使用所有细胞的平均值,这可能适用于包含足够肿瘤和正常细胞的数据集。
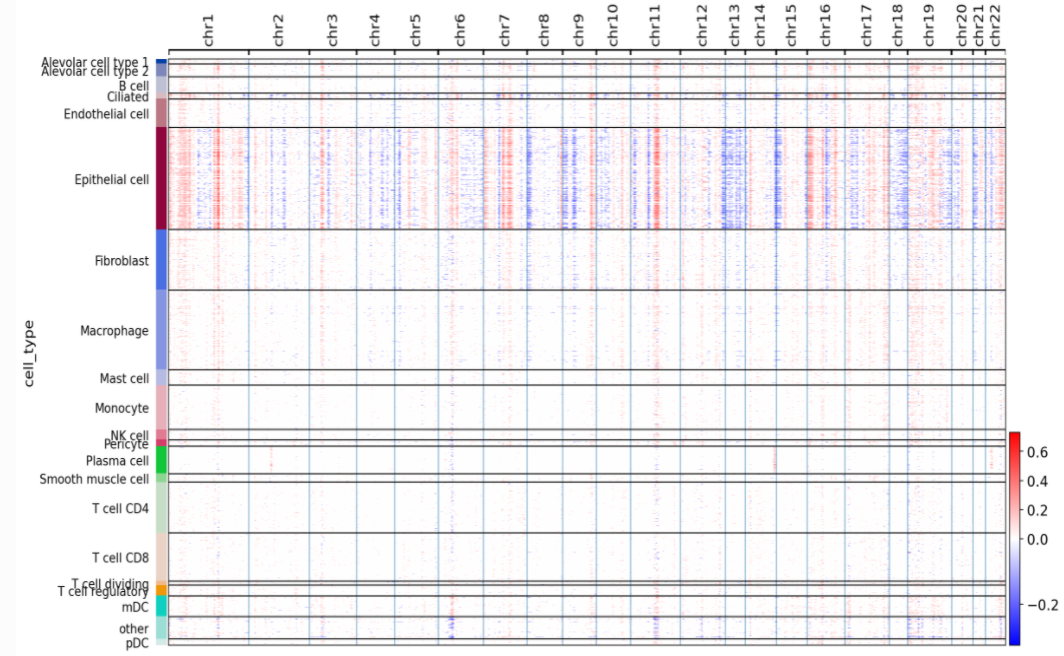
Step3.可视化
绘制热图
现在,可以按细胞类型和染色体绘制平滑的基因表达。可以观察到主要由肿瘤细胞组成的上皮细胞cluster似乎受到拷贝数变化的影响。
cnv.pl.chromosome_heatmap(adata, groupby="cell_type")
CNV聚类和肿瘤细胞鉴定
为了对细胞进行聚类和注释,inferCNVpy镜像了scanpy工作流。以下函数与它们的scanpy对应函数完全一样,只是它们使用CNV剖面矩阵作为输入。使用这些函数,我们可以执行基于UMAP的聚类,并根据CNV数据生成UMAP图。基于这些clusters,我们可以注释肿瘤细胞和正常细胞:

cnv.tl.pca(adata)
cnv.pp.neighbors(adata)
cnv.tl.leiden(adata)
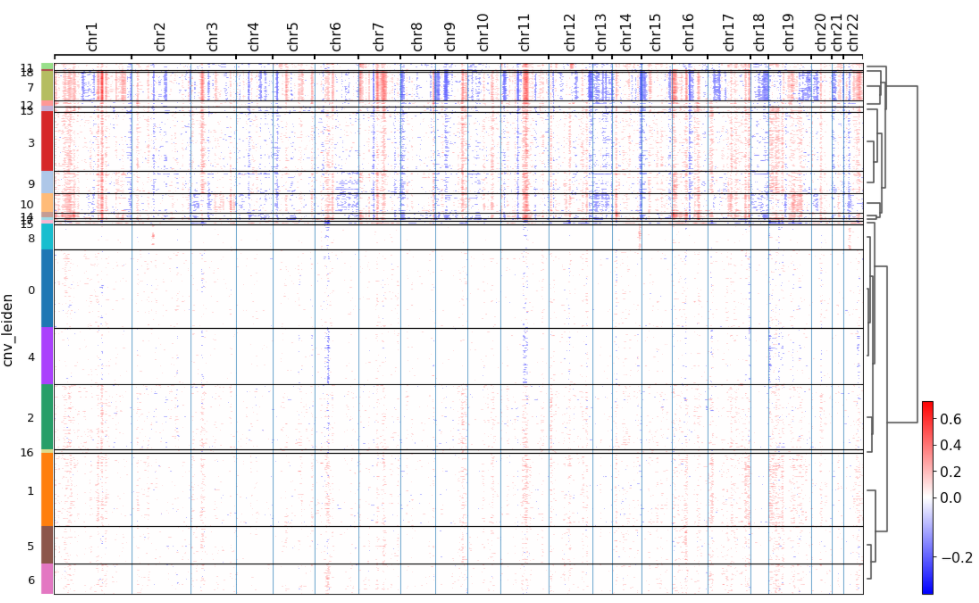
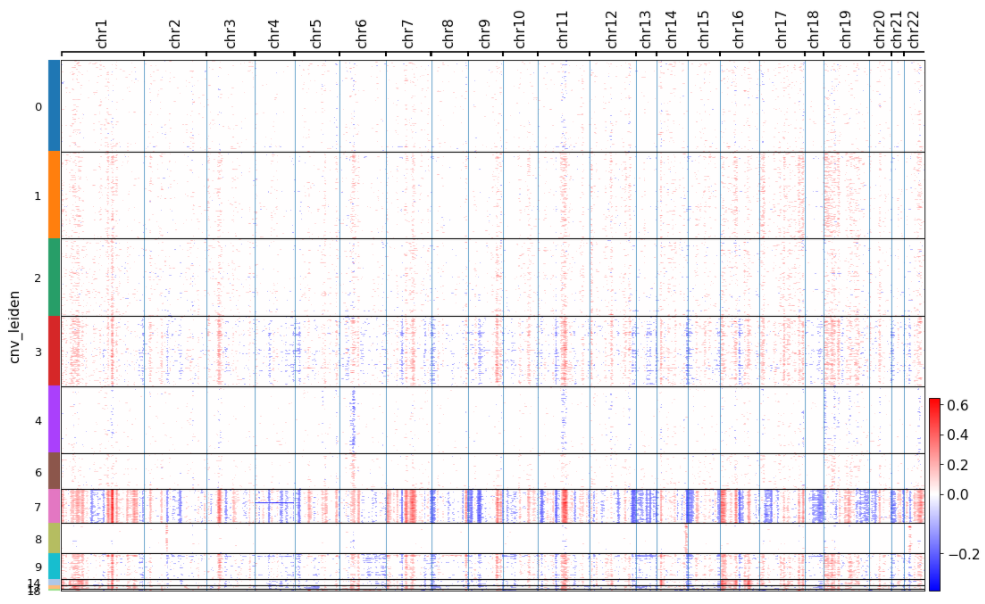
在进行leiden聚类后,我们可以通过CNV聚类来绘制染色体热图。我们可以观察到,与底部的clusters不同,顶部的clusters基本上没有差异表达的基因组区域。差异表达区域可能是由于复制数量的变化,而相应的clusters可能代表肿瘤细胞
cnv.pl.chromosome_heatmap(adata, groupby="cnv_leiden", dendrogram=True)
UMAP plot of CNV profiles
我们可以将相同的clusters可视化为 UMAP 图。此外,infercnvpy.tl.cnv_score() 计算一个汇总分数,量化每个cluster的拷贝数变异量。它被简单地定义为每个cluster的 CNV 矩阵的绝对值的平均值。
cnv.tl.umap(adata)
cnv.tl.cnv_score(adata)
UMAP 图由一大团“正常”细胞和几个具有不同 CNV 分布的较小cluster组成。除了由纤毛细胞组成的cluster“12”外,孤立的cluster都是上皮细胞。这些可能是肿瘤细胞,每个cluster代表一个单独的亚克隆
fig, ((ax1, ax2), (ax3, ax4)) = plt.subplots(2, 2, figsize=(11, 11))
ax4.axis("off")
cnv.pl.umap(
adata,
color="cnv_leiden",
legend_loc="on data",
legend_fontoutline=2,
ax=ax1,
show=False,
)
cnv.pl.umap(adata, color="cnv_score", ax=ax2, show=False)
cnv.pl.umap(adata, color="cell_type", ax=ax3)
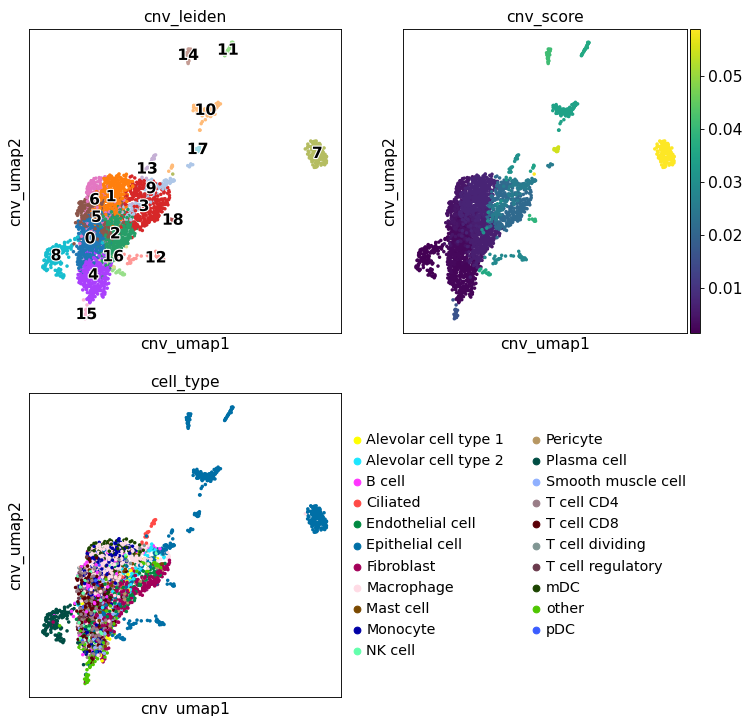
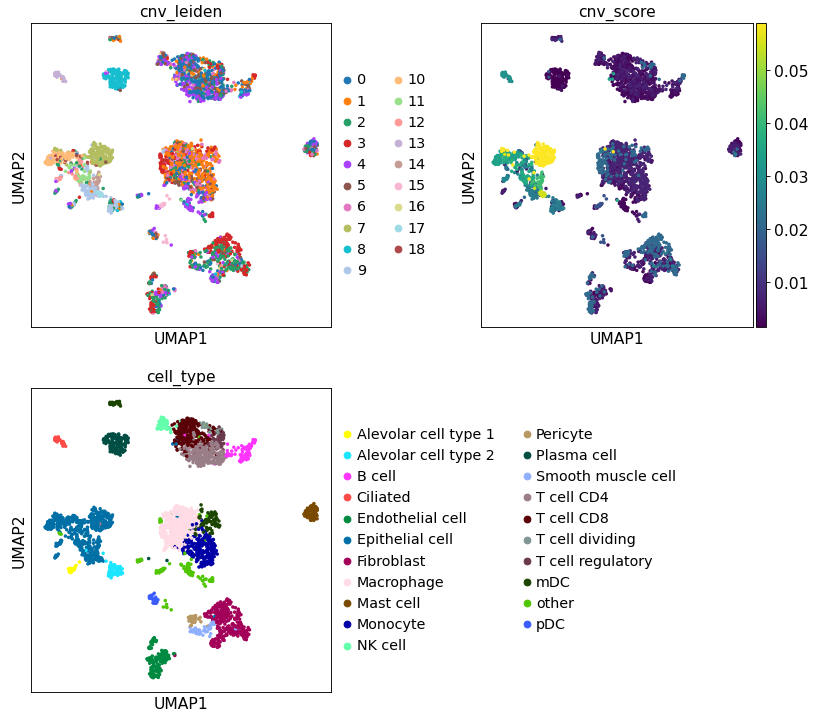
还可以在基于转录组学的 UMAP 图上可视化 CNV 分数和cluster。同样,可以看到存在属于不同 CNV cluster的上皮细胞subcluster,并且这些cluster往往具有最高的 CNV 分数。
fig, ((ax1, ax2), (ax3, ax4)) = plt.subplots(
2, 2, figsize=(12, 11), gridspec_kw=dict(wspace=0.5)
)
ax4.axis("off")
sc.pl.umap(adata, color="cnv_leiden", ax=ax1, show=False)
sc.pl.umap(adata, color="cnv_score", ax=ax2, show=False)
sc.pl.umap(adata, color="cell_type", ax=ax3)
Step5.Classifying tumor cells
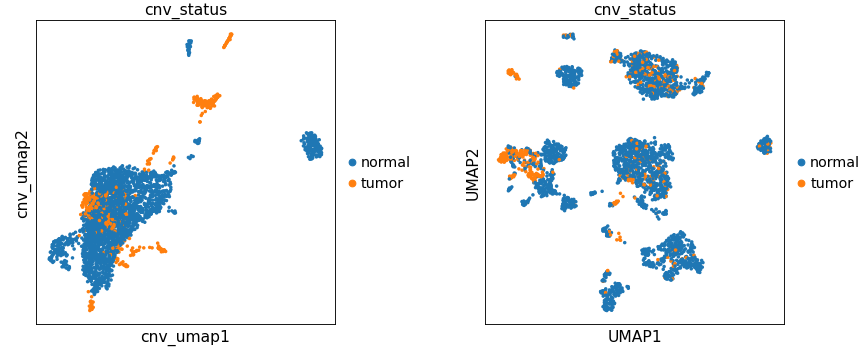
基于这些观察,我们现在可以将细胞分配给“tumor”或“normal”。为此,我们在 adata.obs 中添加一个新列 cnv_status:
adata.obs["cnv_status"] = "normal"
adata.obs.loc[
adata.obs["cnv_leiden"].isin(["10", "13", "15", "5", "11", "16", "12"]), "cnv_status"
] = "tumor"
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(12, 5), gridspec_kw=dict(wspace=0.5))
cnv.pl.umap(adata, color="cnv_status", ax=ax1, show=False)
sc.pl.umap(adata, color="cnv_status", ax=ax2)
cnv.pl.chromosome_heatmap(adata[adata.obs["cnv_status"] == "tumor", :])
 - END -
- END -







