
近日,密歇根大学Goldsmith以及Linic等人在Cell Press细胞出版社期刊Chem Catalysis上发表了一篇新研究,他们使用无监督机器学习的主成分分析法,为开发精准且可解释的材料催化性质的电子-结构描述符提供了一种捷径。该方法有望应用于化学和材料科学交叉领域,并为催化剂的合理设计以及功能材料的构效关系研究提供重要支持。在多相催化领域,电子-结构描述符在催化剂的几何结构和化学吸附性质之间起着重要的联系作用。其中,吸附位点d-投影态密度(DOS)的第一统计矩—d-能带中心,描述了表面位点d-电子态的平均能量,也是目前最流行的用于原子/分子在过渡金属表面上的化学吸附能描述符之一。此外,为进一步提升电子-结构化学吸附描述符在d-能带中心之外的预测能力,一些扩展描述符也被相继开发:比如考虑投影DOS的高阶矩(例如描述d-能带宽度的二阶矩);以及DOS的其他精细结构描述符(例如d-能带相对于费米能级上边缘的位置)等。然而遗憾的是,目前尚没有一个统一的框架可以系统地在一系列不同的材料中准确地识别这些描述符。基于此,本文作者提出了一个数据驱动的工作流,使得电子-结构基化学吸附描述符能够被准确的识别和解释。且进一步,这些电子-结构描述符可以作为桥梁,将催化剂的化学吸附性质与其几何结构和组成联系起来。为此,作者使用主成分分析法(Principal Components Analysis,PCA)来获得材料电子结构的描述符,并利用信号重建来解释每个主成分(PC)描述符捕获的电子结构效应,以及这些效应映射到某一位置几何结构的局部变化(示意图1)。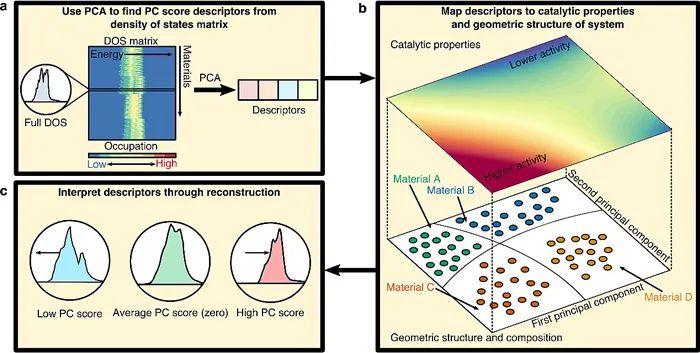
示意图1. 使用PCA自动进行电子结构描述符识别的工作流程。(a)PCA确定DOS矩阵的低维基础(即主成分,PC),以生成PC评分描述符。(b)这些描述符可用于探索材料的电子结构、几何形状和催化性能(如活性)之间的联系。(c)值得注意的是,在每个描述符中捕获的电子-结构效应可以通过DOS重建进行解释分析。具体地,作者主要通过寻找过渡金属合金表面化学吸附的PC描述符证明了这种方法,并进一步通过其与物理基的化学吸附描述符结果进行比对来验证该方法的准确性。本文还拓展了研究金属、金红石型氧化物和钙钛矿型氧化物表面氧活性的方法,并开发了用于量化这些材料氧活性的新型电子-结构描述符。该方法有望在未来应用于物理和化学科学中一些重点材料的电子-结构描述符的开发工作中(例如,固体材料、有机金属催化剂和酶)。作者首先选取了一个具有明确配体和应变效应的层状合金(111)模型体系,以阐明电子结构及几何效应与化学吸附能的相互作用(图1所示)。在层状合金中,配体金属层紧贴于表面之下,而表面金属构成其余部分。本文共考察了245种层状合金:包括Rh合金、Pd合金、Ir合金以及Pt合金。这些被考察的表面金属不仅广泛应用于催化领域且分布于不同的族和周期。配体原子则是根据Hume-Rothery规则和已有的合成报道,选取的具有良好的合金形成特性的金属。此外,应变效应也同样被考虑其中。随后,作者利用Kohn-Sham密度泛函理论(DFT)和PBE泛函模拟了碳、氧、氮和氢原子在层状合金上的化学吸附,且吸附质在应变和非应变层状合金的吸附位点与纯表面金属的吸附位点相同,均为已报道的最稳定位点。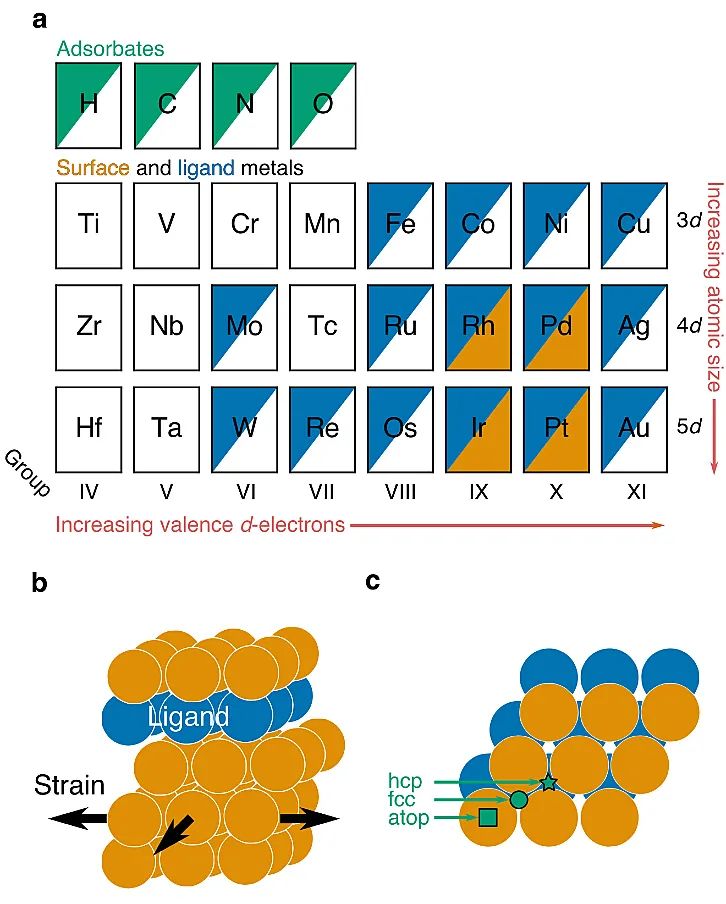
图1. 层状合金的结构和组成。(a)作为表面金属(橙色)、配体金属(蓝色)和吸附剂(绿色)的元素贴图。(b)层状合金模型(111)面。(c)标记吸附位点的层状合金(111)面的俯视图。绿色星表示面心立方(fcc)空位点,绿色圆圈表示六方最密堆积(hcp)空位点,绿色方形表示顶点。主成分分析(PCA)是一种无监督的机器学习技术,它通过将数据投影到一个简化的正交基上(即主成分,PCs)来减少数据集的维数(即描述数据集的变量的数量),以描述数据中的最大方差。这样所有的数据点都将由一组称为PC分数(PC scores)的值来描述,且所得到的PC描述符是与吸附剂无关的合金的电子-结构描述符。与之前将PCA应用于原子、几何或能量特征的ML研究相反,作者将PCA应用于裸催化剂表面的原子投影d-电子DOS。且为了更好的比较三种不同ML模型(PC描述符,传统电子-结构描述符,完整DOS)的性能差异,作者进行了严格的嵌套交叉验证(CV)分析,如图2所示。研究结果表明,使用10个PCs作为输入特征的ML模型的CV均方根误差(RMSEs)与完整DOS模型基本相同,但仅为传统描述符构建模型的一半。此外,在某些回归模型情况下PC描述符模型甚至优于完整DOS模型。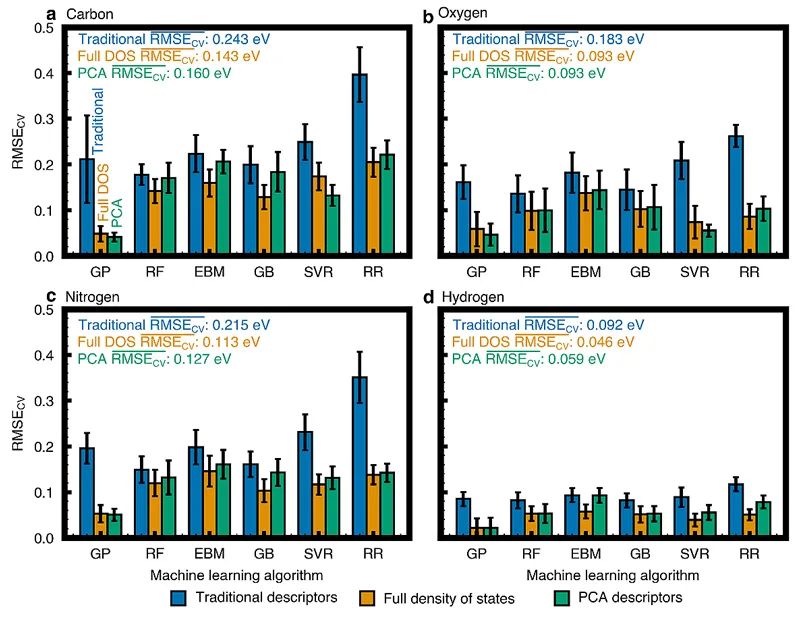
图2. 交叉验证法证实了PCA对不同机器学习算法的普遍性以及预测C(a),O(b),N(c)和H(d)在层状合金上的吸附能。彩色条形图为分别利用传统电子-结构描述符(蓝色)、完整DOS(橙色)以及前十大PC描述符(绿色)构筑的高斯过程回归(GP)、随机森林回归(RF)、可解释增强回归(EBM)、梯度增强回归(GB)、支持向量回归(SVR)以及脊回归(RR)模型的交叉验证误差。将主成分描述符与合金的化学吸附能、几何结构和成分相联系为了分析吸附行为随PC分数的变化趋势,作者生成了前两个PCs的部分依赖图(PDPs),如图3所示。可以看出,随着第一PC分数的增加,四种吸附质的吸附能越高,放热更强。相反,随着第二PC分数的增加,四种吸附质的吸附能变弱。一般说来,PC描述符捕获的电子-结构趋势对不同原子吸附物的吸附过程的影响是相似的。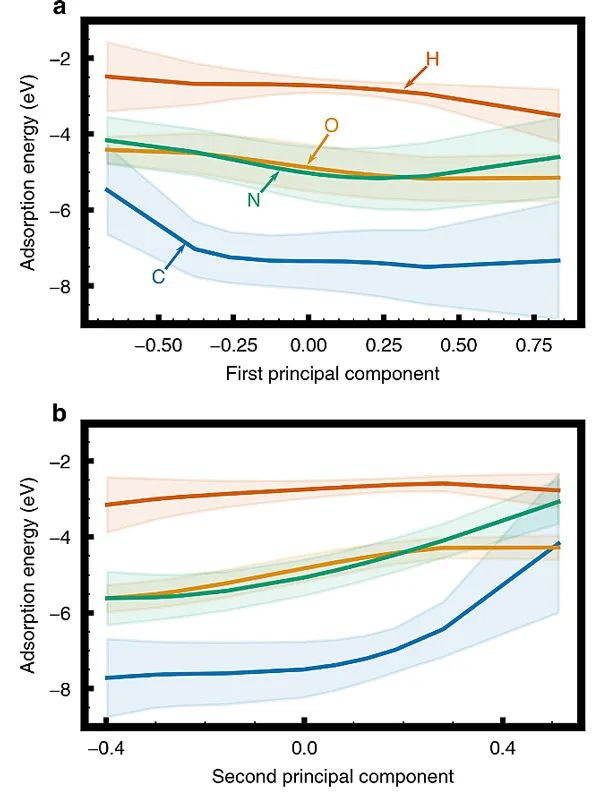
图3. 主成分对C、O、N、H吸附行为的影响。(a)第一和(b)第二主成分的高斯过程回归模型的部分依赖图。为了确定每个PC描述符捕获的电子结构效应,作者还研究了不同PC描述符对DOS重建的函数。图4a和b展现了第一和第二PC对DOS重建的影响。而图4c-f则显示了第一和第二PC对DOS重构的统计矩(d-能带中心/第一矩,宽度/第二矩,偏斜度/第三矩、峰度/第四矩)相关的汇总统计行为的影响。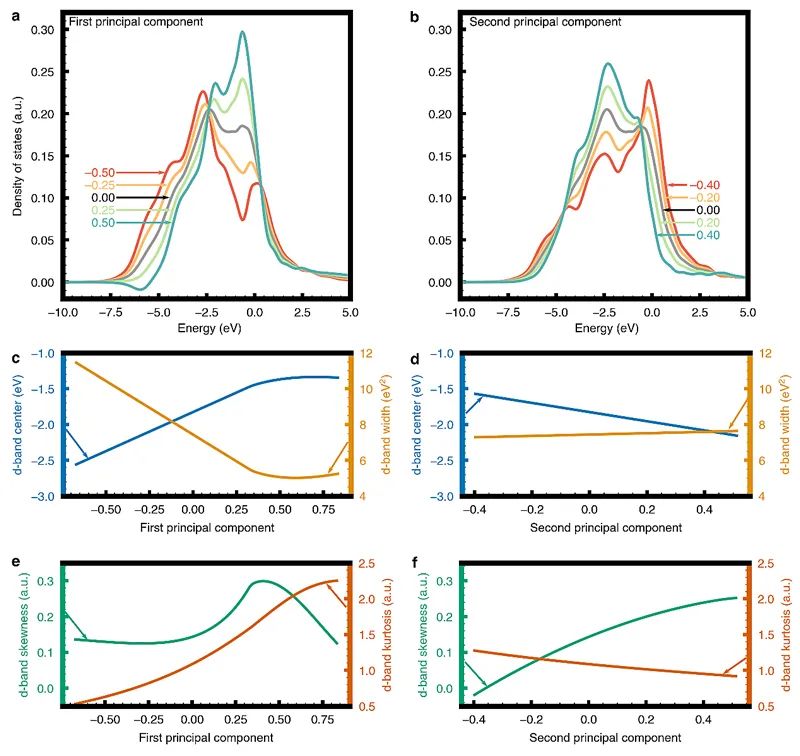
如何超越电子结构模型,将合金的几何结构和组成与其化学吸附强度联系起来,对于建立可预测的几何-化学吸附强度模型至关重要。而电子结构作为几何结构和化学吸附之间的重要纽带,可以帮助识别几何描述符。有鉴于此,作者通过箱型图和锋群图对第一和第二PC分数分布,以及电子结构随合金几何结构和组成变化情况进行了研究,如图5所示。作者首先分析了PC分数和合金表面金属之间的关系(图5a,b),研究结果表明,对于有着相似的d-价电子结构的5d和4d表面金属合金,具有更大原子半径的5d表面金属合金与吸附质的结合力更弱,即表面金属的尺寸大小为影响其表面几何效应的第一个PC分数。而第二个PC分数主要取决于表面金属中d-价电子的数量(图5b),即具有更多d-价电子的过渡金属吸附强度较弱。图5c,d则显示了次表面配体原子对第一和第二PC分数的影响。图5c表明,具有后过渡金属的合金表面展现出更高的第一PC分数(以及更强的化学吸附),图5d则表明,第二PC与配体金属特性之间无明显关联。进一步,作者利用PC描述符分析了几何应变对电子结构和化学吸附强度的影响(图5e,f):应变对第一PC评分有显著影响,但几乎不会影响第二PC分数。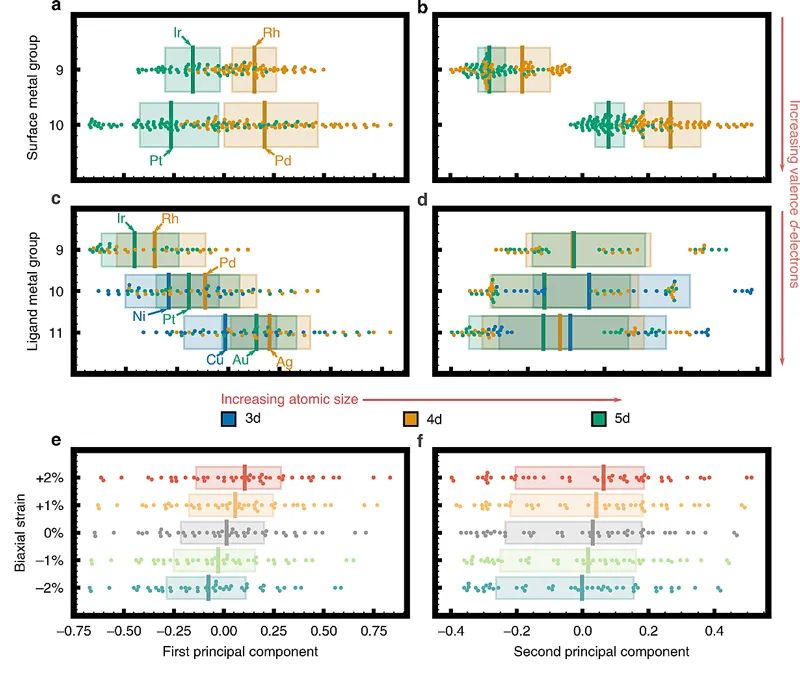
示意图2则进一步对上述结果进行了总结,即d-价电子数量和金属原子的尺寸是控制化学吸附强度的两个关键参数。一般来说,增加配体和表面金属的尺寸,减小配体金属中d-价电子的数量,对应第一PC的减少和化学吸附减弱;而表面d-电子数量的增加则对应于第二PC的增加和化学吸附减弱。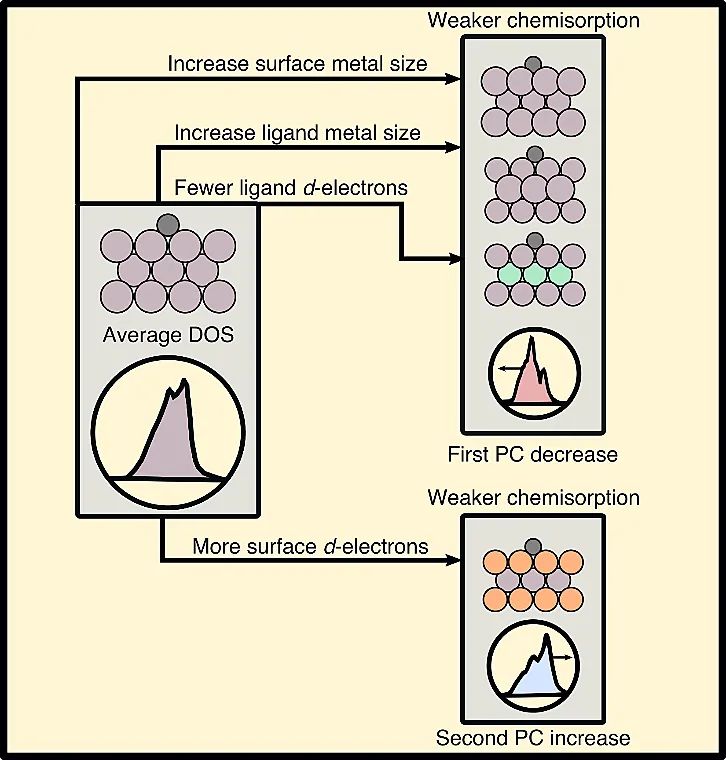
示意图2. 表面和配体金属的性质对合金化学吸附强度的影响。利用PCA寻找金属和金属氧化物氧活性的电子结构描述符
寻找活性氧物种的电子-结构描述符一直是一个长期存在的挑战,对于理解材料的化学催化氧化活性如甲烷的氧化偶联、析氧反应、丙烯环氧化等具有重要意义。因此,作者将PCA进一步应用于金属、金红石型氧化物以及钙钛矿型氧化物(ABO3)上的表面氧物种的DOS,构建了O 2p态的描述符,并对其表面氧的O 2p键合和反键合轨道随第一PC分数的变化函数(图6b),第一PC分数对表面氧活性影响的部分依赖曲线(图6c)以及第一PC分数随基础金属周期(图6d)和主族(图6e)变化函数进行了研究。通过重建对第一PC描述符的分析表明,该描述符捕获的趋势与键的物理化学描述是一致的;而相对于PC描述符的表面氧活性趋势表明,第一PC分数高的材料,其表面氧种类不稳定,反键轨道的相对填充量较高,氧化活性较强。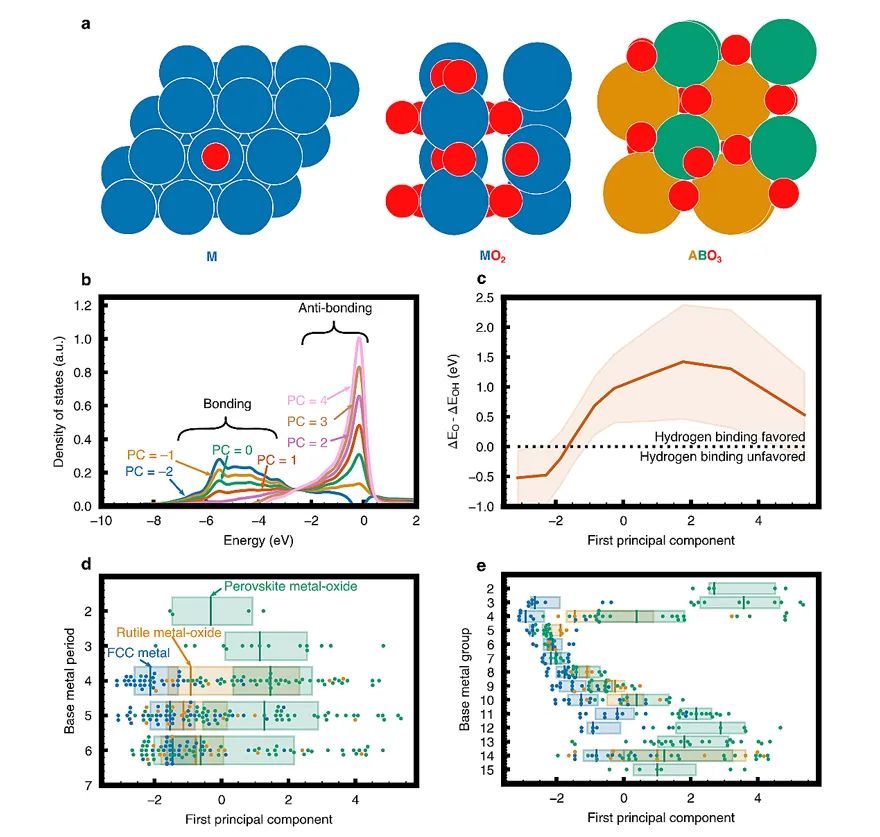
图6. 基于PCA的金属和金属氧化物氧活性的电子-结构描述符。无监督机器学习是一种前景广阔的用于开发材料描述符的有效途径。本文利用简单的主成分分析法(PCA)发展了基于后过渡金属合金d-能带投影态密度的电子-结构描述符。其建立的PC描述符ML模型可精确预测化学吸附,且性能优于使用传统电子-结构描述符构建的模型。更重要的是,这种PC描述符是可解释的,从而可以深入了解材料的电子结构如何与表面几何形状及组成相连接,并最终达到化学吸附强度。本文通过考察金属、金红石型氧化物和钙钛矿型氧化物的表面氧活性,证实了该方法对于不同催化系统的普适性。未来,该方法有望拓展到其他催化系统,如金属间和随机合金、氮化物以及硫化物,以及其他跨化学和材料科学的应用领域中。Uncovering electronic and geometric descriptors of chemical activity for metal alloys and oxides using unsupervised machine learningJacques A. Esterhuizen, Bryan R. Goldsmith, Suljo LinicChem Catal., 2021, DOI: 10.1016/j.checat.2021.07.014

点击“阅读原文”,查看 化学 • 材料 领域所有收录期刊










